#Oxidative phosphorylation (OXPHOS)
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
In February 2021, Tumblr had 518.6 million blog accounts.
Text
The Role of Mitochondria in Prader–Willi Syndrome
1. Introduction
Prader–Willi Syndrome (PWS) is a rare genetic disorder resulting from the lack of expression of paternally inherited genes in the 15q11–q13 chromosomal region. Clinically, it is characterized by neonatal hypotonia, hyperphagia, obesity, short stature, cognitive impairment, hypogonadism, and behavioral issues. Historically, these features have been attributed to hypothalamic dysfunction. However, recent research highlights a significant role of mitochondrial dysfunction in the metabolic and neuromuscular symptoms of PWS.
2. Mitochondrial Function and Its Systemic Relevance
Mitochondria are cellular organelles essential for energy production through oxidative phosphorylation (OXPHOS). They also regulate reactive oxygen species (ROS) generation, calcium signaling, and apoptosis. In energy-demanding tissues such as brain and muscle, mitochondrial integrity is vital. Any impairment in mitochondrial function disrupts cellular energy metabolism, often resulting in clinical features seen in syndromes like PWS.
3. Bioenergetic Deficits in PWS
Patients with PWS exhibit symptoms like muscle weakness, reduced endurance, and fatigue—all suggestive of compromised mitochondrial energy production. Cellular studies on fibroblasts derived from PWS individuals have shown decreased basal respiration, reduced ATP production, and limited spare respiratory capacity. These deficits indicate impaired mitochondrial oxidative phosphorylation and diminished cellular energy reserves.
4. Electron Transport Chain Abnormalities
Specific defects in the electron transport chain (ETC), particularly in Complex I, have been reported in PWS. Complex I initiates the ETC by transferring electrons from NADH to ubiquinone. Defects in Complex I result in lower ATP generation and an increase in ROS. The resultant oxidative stress can damage mitochondrial DNA, lipids, and proteins, further impairing mitochondrial function and exacerbating clinical symptoms.
5. Coenzyme Q10 Deficiency
Coenzyme Q10 (CoQ10) is a lipid-soluble molecule vital for electron transport between Complexes I/II and III. It also acts as an antioxidant, protecting membranes and cellular structures from oxidative damage. In individuals with PWS, CoQ10 levels are often significantly lower than in the general population. This deficiency disrupts electron flow, reduces ATP synthesis, and increases oxidative stress. Clinically, CoQ10 deficiency may contribute to hypotonia, poor endurance, and delayed developmental milestones in PWS patients.
6. Fatty Acid Oxidation and Acylcarnitine Abnormalities
In PWS, metabolic profiling has revealed elevated acylcarnitine levels, particularly medium- and short-chain species. These findings suggest a disruption in fatty acid β-oxidation, a key mitochondrial process. Accumulated acylcarnitines are indicative of incomplete fatty acid utilization, which may stem from defective carnitine transport or mitochondrial enzyme activity. As fatty acids are critical energy substrates during fasting and exercise, their impaired oxidation contributes to energy failure and obesity in PWS.
7. Carnitine Deficiency and Transport Impairment
Carnitine is essential for the transport of long-chain fatty acids into mitochondria for β-oxidation. Some studies have reported reduced serum carnitine levels in individuals with PWS, especially in infants and young children. Carnitine deficiency may result from reduced intake, increased renal losses, or altered synthesis. Supplementation with carnitine has been associated with improvements in muscle tone and energy levels in some cases, suggesting its therapeutic potential.
8. Gene Expression and Mitochondrial Regulation
PWS results from the loss of paternal expression of genes in the 15q11–q13 region, including small nucleolar RNAs (snoRNAs) and non-coding RNAs involved in RNA processing and regulation. Transcriptomic studies in mouse models have shown dysregulation of genes associated with mitochondrial function, including those involved in ribosomal assembly, fatty acid metabolism, and oxidative phosphorylation. These molecular alterations reinforce the hypothesis that mitochondrial dysfunction is a primary contributor to the PWS phenotype.
9. Structural Mitochondrial Alterations
Electron microscopy studies in animal models of PWS have demonstrated mitochondrial structural abnormalities, including swelling, disorganized cristae, and altered mitochondrial number. These findings correlate with decreased efficiency of oxidative metabolism and increased oxidative damage. Mitochondrial remodeling in cardiac, neural, and skeletal muscle tissues may underlie systemic features such as cardiomyopathy, cognitive deficits, and fatigue.
10. Therapeutic Implications
Understanding mitochondrial dysfunction in PWS opens the door to targeted therapies. The following strategies are under consideration:
Coenzyme Q10 Supplementation: Administered to enhance electron transport and reduce oxidative stress. Anecdotal reports have shown improved motor function and alertness in children receiving CoQ10.
Carnitine Therapy: May support fatty acid transport and improve energy production. Used in cases with documented deficiency or fatigue.
Antioxidants: Agents such as alpha-lipoic acid, vitamin E, or NAC might mitigate ROS-related damage and preserve mitochondrial integrity.
Mitochondrial Biogenesis Enhancers: Agents that stimulate mitochondrial replication and function, such as PGC-1α activators, are under investigation.
Metabolic Monitoring: Regular assessment of acylcarnitine profiles, lactate, and oxidative stress markers can help personalize treatment.
11. Future Directions
To advance clinical care for PWS, several research priorities have emerged:
Controlled Clinical Trials: Rigorous evaluation of CoQ10 and carnitine supplementation is needed to assess efficacy and safety.
Multi-Tissue Profiling: Comprehensive mitochondrial function studies in muscle, brain, liver, and adipose tissues will clarify tissue-specific vulnerabilities.
Genotype–Phenotype Correlation: Understanding how specific genetic deletions affect mitochondrial pathways can guide personalized interventions.
Biomarker Development: Identifying mitochondrial biomarkers in blood or urine could enable early detection of dysfunction and monitoring of treatment response.
12. Conclusion
While traditionally attributed to hypothalamic dysfunction, Prader–Willi syndrome also involves systemic mitochondrial impairment. Defects in energy metabolism, fatty acid oxidation, and antioxidant defense converge to produce many of the syndrome's characteristic features. Recognition of mitochondrial involvement in PWS pathophysiology has the potential to refine diagnosis, improve symptom management, and inspire new therapeutic avenues. Future research integrating genomics, bioenergetics, and clinical studies will be essential in translating this understanding into effective patient care.
#Prader–Willi Syndrome#Mitochondrial dysfunction#Oxidative phosphorylation (OXPHOS)#Coenzyme Q10 (CoQ10)#Carnitine deficiency#Electron transport chain#Complex I activity#Fatty acid oxidation#Acylcarnitines#Reactive oxygen species (ROS)#Mitochondrial bioenergetics#Energy metabolism#Hypotonia in PWS#Metabolic profiling#Mitochondrial gene expression#Mitochondrial structure#Mitochondrial therapy#Antioxidant supplementation#Mitochondrial biogenesis#Neuromuscular symptoms PWS
0 notes
Text
Story at-a-glance
Suppression of mitochondrial ATP production prevents apoptosis and activates the NLRP3 inflammasome, a key player in inflammation and disease
Inhibitors of oxidative phosphorylation (OXPHOS) lead to changes in mitochondrial cristae structure and retention of cytochrome c, which is necessary for NLRP3 activation but not sufficient on its own
Activation of the NLRP3 inflammasome requires two signals, one of which is mitochondrial, highlighting the complexity of its regulation
Diverse NLRP3 activators share the ability to suppress apoptosis, allowing damaged cells to survive and contributing to chronic inflammation and cancer
Mitochondrial dysfunction is closely linked to inflammation and various diseases, emphasizing the importance of understanding these mechanisms for optimal health
7 notes
·
View notes
Text
4 notes
·
View notes
Text
Study uncovers the molecular evolutionary strategies of the OxPhos system
Mitochondria are the body’s “energy factories,” and their proper function is essential for life. Inside mitochondria, a set of complexes called the oxidative phosphorylation (OxPhos) system acts like a biochemical assembly line, transforming oxygen and nutrients into usable energy. Now, the study, led by the GENOXPHOS group at the Spanish National Centre for Cardiovascular Research (CNIC) and the…
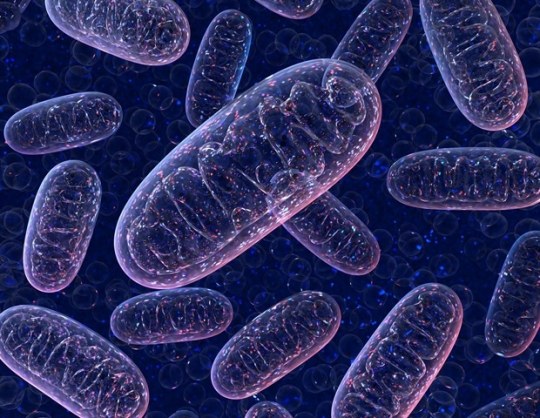
View On WordPress
0 notes
Text
SMART researchers develop a method to enhance effectiveness of cartilage repair therapy
New Post has been published on https://sunalei.org/news/smart-researchers-develop-a-method-to-enhance-effectiveness-of-cartilage-repair-therapy/
SMART researchers develop a method to enhance effectiveness of cartilage repair therapy

Researchers from the Critical Analytics for Manufacturing Personalized-Medicine (CAMP) interdisciplinary research group at the Singapore-MIT Alliance for Research and Technology (SMART), MIT’s research enterprise in Singapore, alongside collaborators from the National University of Singapore Tissue Engineering Programme, have developed a novel method to enhance the ability of mesenchymal stromal cells (MSCs) to generate cartilage tissue by adding ascorbic acid during MSC expansion. The research also discovered that micro-magnetic resonance relaxometry (µMRR), a novel process analytical tool developed by SMART CAMP, can be used as a rapid, label-free process-monitoring tool for the quality expansion of MSCs.
Articular cartilage, a connective tissue that protects the bone ends in joints, can degenerate due to injury, age, or arthritis, leading to significant joint pain and disability. Especially in countries — such as Singapore — that have an active, aging population, articular cartilage degeneration is a growing ailment that affects an increasing number of people. Autologous chondrocyte implantation is currently the only Food and Drug Administration-approved cell-based therapy for articular cartilage injuries, but it is costly, time-intensive, and requires multiple treatments. MSCs are an attractive and promising alternative as they have shown good safety profiles for transplantation. However, clinical use of MSCs is limited due to inconsistent treatment outcomes arising from factors such as donor-to-donor variability, variation among cells during cell expansion, and non-standardized MSC manufacturing protocols.
The heterogeneity of MSCs can lead to variations in their biological behavior and treatment outcomes. While large-scale MSC expansions are required to obtain a therapeutically relevant number of cells for implantation, this process can introduce cell heterogeneity. Therefore, improved processes are essential to reduce cell heterogeneity while increasing donor cell numbers with improved chondrogenic potential — the ability of MSCs to differentiate into cartilage cells to repair cartilage tissue — to pave the way for more effective and consistent MSC-based therapies.
In a paper titled “Metabolic modulation to improve MSC expansion and therapeutic potential for articular cartilage repair,” published in the scientific journal Stem Cell Research and Therapy, CAMP researchers detailed their development of a priming strategy to enhance the expansion of quality MSCs by modifying the way cells utilize energy. The research findings have shown a positive correlation between chondrogenic potential and oxidative phosphorylation (OXPHOS), a process that harnesses the reduction of oxygen to create adenosine triphosphate — a source of energy that drives and supports many processes in living cells. This suggests that manipulating MSC metabolism is a promising strategy for enhancing chondrogenic potential.
Using novel PATs developed by CAMP, the researchers explored the potential of metabolic modulation in both short- and long-term harvesting and reseeding of cells. To enhance their chondrogenic potential, they varied the nutrient composition, including glucose, pyruvate, glutamine, and ascorbic acid (AA). As AA is reported to support OXPHOS and its positive impact on chondrogenic potential during differentiation — a process in which immature cells become mature cells with specific functions — the researchers further investigated its effects during MSC expansion.
The addition of AA to cell cultures for one passage during MSC expansion and prior to initiation of differentiation was found to improve chondrogenic differentiation, which is a critical quality attribute (CQA) for better articular cartilage repair. Longer-term AA treatment led to a more than 300-fold increase in the yield of MSCs with enhanced chondrogenic potential, and reduced cell heterogeneity and cell senescence — a process by which a cell ages and permanently stops dividing but does not die — when compared to untreated cells. AA-treated MSCs with improved chondrogenic potential showed a robust shift in metabolic profile to OXPHOS. This metabolic change correlated with μMRR measurements, which helps identify novel CQAs that could be implemented in MSC manufacturing for articular cartilage repair.
The research also demonstrates the potential of the process analytical tool developed by CAMP, micromagnetic resonance relaxometry (μMRR) — a miniature benchtop device that employs magnetic resonance imaging (MRI) imaging on a microscopic scale — as a process-monitoring tool for the expansion of MSCs with AA supplementation. Originally used as a label-free malaria diagnosis method due to the presence of paramagnetic hemozoin particles, μMRR was used in the research to detect senescence in MSCs. This rapid, label-free method requires only a small number of cells for evaluation, which allows for MSC therapy manufacturing in closed systems — a system for protecting pharmaceutical products by reducing contamination risks from the external environment — while enabling intermittent monitoring of a limited lot size per production.
“Donor-to-donor variation, intrapopulation heterogeneity, and cellular senescence have impeded the success of MSCs as a standard of care therapy for articular cartilage repair. Our research showed that AA supplementation during MSC expansion can overcome these bottlenecks and enhance MSC chondrogenic potential,” says Ching Ann Tee, senior postdoc at SMART CAMP and first author of the paper. “By controlling metabolic conditions such as AA supplementation, coupled with CAMP’s process analytical tools such as µMRR, the yield and quality of cell therapy products could be significantly increased. This breakthrough could help make MSC therapy a more effective and viable treatment option and provide standards for improving the manufacturing pipeline.”
“This approach of utilizing metabolic modulation to improve MSC chondrogenic potential could be adapted into similar concepts for other therapeutic indications, such as osteogenic potential for bone repair or other types of stem cells. Implementing our findings in MSC manufacturing settings could be a significant step forward for patients with osteoarthritis and other joint diseases, as we can efficiently produce large quantities of high-quality MSCs with consistent functionality and enable the treatment of more patients,” adds Professor Laurie A. Boyer, principal investigator at SMART CAMP, professor of biology and biological engineering at MIT, and corresponding author of the paper.
The research is conducted by SMART and supported by the National Research Foundation Singapore under its Campus for Research Excellence and Technological Enterprise program.
0 notes
Quote
Brain metastasis of advanced breast cancer often results in deleterious consequences. Metastases to the brain lead to significant challenges in treatment options, as the blood–brain barrier (BBB) prevents conventional therapy. Thus, we hypothesized that creation of a nanoparticle (NP) that distributes to both primary tumor site and across the BBB for secondary brain tumor can be extremely beneficial. Here, we report a simple targeting strategy to attack both the primary breast and secondary brain tumors utilizing a single NP platform. The nature of these mitochondrion-targeted, BBB-penetrating NPs allow for simultaneous targeting and drug delivery to the hyperpolarized mitochondrial membrane of the extracranial primary tumor site in addition to tumors at the brain. By utilizing a combination of such dual anatomical distributing NPs loaded with therapeutics, we demonstrate a proof-of-concept idea to combat the increased metabolic plasticity of brain metastases by lowering two major energy sources, oxidative phosphorylation (OXPHOS) and glycolysis. By utilizing complementary studies and genomic analyses, we demonstrate the utility of a chemotherapeutic prodrug to decrease OXPHOS and glycolysis by pairing with a NP loaded with pyruvate dehydrogenase kinase 1 inhibitor. Decreasing glycolysis aims to combat the metabolic flexibility of both primary and secondary tumors for therapeutic outcome. We also address the in vivo safety parameters by addressing peripheral neuropathy and neurobehavior outcomes. Our results also demonstrate that this combination therapeutic approach utilizes mitochondrial genome targeting strategy to overcome DNA repair–based chemoresistance mechanisms.
Simultaneous targeting of peripheral and brain tumors with a therapeutic nanoparticle to disrupt metabolic adaptability at both sites | PNAS
0 notes
Text
Cells, Vol. 12, Pages 2313: Let-7g Upregulation Attenuated the KRAS–PI3K–Rac1–Akt Axis-Mediated Bioenergetic Functions
The aberrant activation of signaling pathways contributes to #cancer cells with metabolic reprogramming. Thus, targeting signaling modulators is considered a potential therapeutic strategy for #cancer. Subcellular fractionation, coimmunoprecipitation, biochemical analysis, and gene manipulation experiments revealed that decreasing the interaction of kirsten rat sarcoma viral oncogene homolog (KRAS) with p110α in lipid rafts with the use of naringenin (NGN), a citrus flavonoid, causes lipid raft-associated phosphatidylinositol 3-kinase (PI3K)GTP-ras-related C3 botulinum toxin substrate 1 (Rac1)protein kinase B (Akt)-regulated metabolic dysfunction of glycolysis and mitochondrial oxidative phosphorylation (OXPHOS), leading to apoptosis in human nasopharyngeal carcinoma (NPC) cells. The use of lethal-7g (let-7g) mimic and let-7g inhibitor confirmed that elevated let-7g resulted in a decrease in KRAS expression, which attenuated the PI3KRac1AktBCL-2/BCL-xL-modulated mitochondrial energy metabolic functions. Increased let-7g depends on the suppression of the #RNA-specificity of monocyte chemoattractant protein-induced protein-1 (MCPIP1) ribonuclease since NGN specifically blocks the degradation of pre-let-7g by NPC cell-derived immunoprecipitated MCPIP1. Converging lines of evidence indicate that the inhibition of MCPIP1 by NGN leads to let-7g upregulation, suppressing oncogenic KRAS-modulated PI3K–Rac1–Akt signaling and thereby impeding the metabolic activities of aerobic glycolysis and mitochondrial OXPHOS. https://www.mdpi.com/2073-4409/12/18/2313?utm_source=dlvr.it&utm_medium=tumblr
0 notes
Text
Mitochondrial Dysfunction in SLC6A1: A Molecular and Cellular Perspective
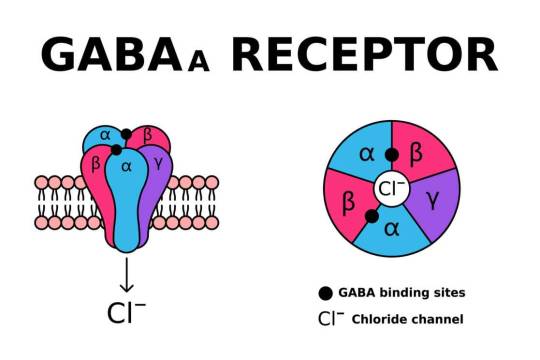
SLC6A1 encodes the gamma-aminobutyric acid (GABA) transporter type 1 (GAT1), a crucial component of inhibitory neurotransmission. Pathogenic variants in SLC6A1 lead to neurological disorders, primarily epilepsy, developmental delay, and neuropsychiatric conditions. While its role in GABAergic signaling is well established, emerging evidence suggests an intersection with mitochondrial dysfunction, which exacerbates disease pathology. This article explores the molecular and cellular mechanisms linking SLC6A1 mutations to mitochondrial impairment, highlighting alterations in energy metabolism, oxidative stress, and mitochondrial dynamics.
1. Introduction The SLC6A1 gene encodes the GAT1 transporter, responsible for reuptaking GABA from the synaptic cleft into presynaptic neurons and astrocytes. Disruptions in SLC6A1 impair inhibitory neurotransmission, contributing to hyperexcitability in neuronal circuits. Recent studies indicate a link between SLC6A1 dysfunction and mitochondrial abnormalities, underscoring a metabolic component to disease pathogenesis. The mitochondrial connection is crucial as these organelles regulate neuronal energy homeostasis and apoptosis. Understanding these mechanisms is essential for dissecting the full scope of SLC6A1-related disorders.
2. Role of SLC6A1 in Cellular and Mitochondrial Function Neurons exhibit high metabolic demand, relying heavily on mitochondria for adenosine triphosphate (ATP) production. GABA metabolism interfaces with mitochondrial pathways, influencing oxidative phosphorylation (OXPHOS) and redox balance. SLC6A1 mutations impair GABA uptake, potentially disrupting mitochondrial function through dysregulated Krebs cycle activity, altered ATP synthesis, and excessive reactive oxygen species (ROS) generation. Additionally, GABAergic dysfunction affects calcium signaling, further impacting mitochondrial integrity.
3. Energy Metabolism and ATP Production Mitochondria generate ATP primarily through OXPHOS. Deficient GABA uptake alters cellular excitability, increasing ATP demand while simultaneously impairing ATP synthesis. Studies show that neurons with SLC6A1 mutations exhibit reduced mitochondrial membrane potential (∆ψm), leading to inefficient ATP generation. Moreover, compensatory glycolysis often fails to meet neuronal energy demands, resulting in cellular stress and neuronal dysfunction.
4. Oxidative Stress and ROS Dysregulation Mitochondria are primary sites of ROS production, which serve as signaling molecules in normal physiology but become deleterious when unregulated. SLC6A1 mutations contribute to ROS imbalance, leading to oxidative stress and lipid peroxidation. Elevated ROS levels have been reported in neurons with impaired GABAergic signaling, suggesting that SLC6A1 mutations exacerbate mitochondrial oxidative damage. This process triggers mitochondrial DNA (mtDNA) mutations, protein oxidation, and lipid peroxidation, further compromising mitochondrial integrity.
5. Calcium Homeostasis and Mitochondrial Dysfunction Neuronal activity depends on tightly regulated calcium homeostasis. Mitochondria buffer intracellular calcium, maintaining synaptic function and preventing excitotoxicity. SLC6A1 dysfunction alters calcium flux due to disrupted GABAergic inhibition, leading to excessive mitochondrial calcium uptake. This triggers the mitochondrial permeability transition pore (mPTP), resulting in bioenergetic failure and apoptotic signaling cascades. Elevated cytosolic calcium further dysregulates mitochondrial enzyme activity, exacerbating metabolic dysfunction.
6. Mitochondrial Dynamics and Biogenesis Mitochondria undergo continuous fission and fusion to adapt to cellular demands. Impaired mitochondrial dynamics are observed in neurons harboring SLC6A1 mutations, leading to fragmented and dysfunctional mitochondria. The fusion-fission imbalance results in defective mitochondrial quality control, accumulation of damaged organelles, and impaired biogenesis. Downregulation of mitophagy-related proteins such as PINK1 and Parkin has been documented in models of SLC6A1 dysfunction, suggesting defective clearance of impaired mitochondria.
7. Synaptic Dysfunction and Mitochondrial Interactions Neurotransmission relies on synaptic mitochondria to meet localized energy demands. GABAergic synapses, in particular, require significant mitochondrial support due to their reliance on ATP-dependent vesicular transport and receptor function. SLC6A1 mutations disrupt synaptic mitochondrial positioning, reducing ATP availability at synapses. This impairment contributes to synaptic dysfunction, decreased inhibitory tone, and aberrant excitatory-inhibitory balance, which are hallmarks of SLC6A1-related neurological disorders.
8. Neuroinflammation and Mitochondrial Dysfunction Mitochondria modulate immune responses through ROS production and inflammatory cytokine signaling. Neurons with SLC6A1 mutations exhibit increased inflammatory markers, such as interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α), indicative of neuroinflammation. Mitochondrial dysfunction exacerbates this process by activating microglia and astrocytes, leading to chronic neuroinflammatory states. This further damages neuronal mitochondria, perpetuating a vicious cycle of dysfunction and degeneration.
9. Genetic and Epigenetic Influences on Mitochondrial Dysfunction Mutations in SLC6A1 not only affect protein function but also influence mitochondrial gene expression and epigenetics. Studies indicate altered expression of nuclear-encoded mitochondrial genes, including those involved in OXPHOS. Additionally, epigenetic modifications such as DNA methylation and histone acetylation impact mitochondrial biogenesis and function in SLC6A1-related disorders. Dysregulated mitochondrial gene transcription exacerbates bioenergetic failure, compounding neurological deficits.
10. Conclusion Mitochondrial dysfunction is an emerging pathological mechanism in SLC6A1-related disorders, contributing to energy deficits, oxidative stress, impaired calcium homeostasis, defective mitochondrial dynamics, and synaptic dysfunction. Understanding the interplay between SLC6A1 mutations and mitochondrial abnormalities provides insights into disease pathogenesis, paving the way for targeted metabolic and neuroprotective interventions. Future research should focus on elucidating the precise molecular pathways linking SLC6A1 dysfunction to mitochondrial pathology, ultimately aiding in the development of novel therapeutic strategies.
#SLC6A1 gene#Mitochondrial dysfunction#GABA transporter (GAT1)#Neurological disorders#Oxidative stress#Mitochondrial energy metabolism#ATP production#Reactive oxygen species (ROS)#Mitochondrial membrane potential#Calcium homeostasis#Neuronal excitability#Mitochondrial biogenesis#Mitochondrial dynamics#Synaptic dysfunction#Neuroinflammation#Mitochondrial quality control#Mitochondrial permeability transition pore (mPTP)#Neurodegeneration#Epigenetic modifications in mitochondria#Mitochondrial oxidative phosphorylation (OXPHOS)
0 notes
Text
New Ivermectin, Fen Ben Combination Therapy Shows Promise
George Webb
Oct 17, 2024
A new scientific paper has been published in the Journal of Orthomolecular Medicine on the benefits of Ivermectin and Fen Ben in combination with other effective drugs against cancer.
Here is the ChatGPT 4.0 Summary
Key points:
Mitochondrial impairment is central to cancer initiation, making it a critical therapeutic target.
Cancer cells heavily depend on glucose and glutamine due to dysfunctional mitochondria.
Vitamin C shows significant cytotoxic effects against cancer cells and reduces metastasis.
Vitamin D enhances mitochondrial function and reduces cancer incidence and metastasis.
Zinc protects mitochondria from oxidative damage and enhances apoptosis in cancer cells.
Ivermectin induces autophagy and apoptosis of cancer cells, showing efficacy in reducing tumor size.
Fasting and ketogenic diets are effective in inhibiting the primary energy pathways that cancer cells rely on.
Exercise increases mitochondrial respiration and can inhibit cancer progression.
Hyperbaric oxygen therapy (HBOT) works synergistically with ketogenic diets to suppress tumor growth.The paper centers around a process called Oxidative Phosphorylation, which is abbreviated as OxPhos.
1 note
·
View note
Text
more from Doris Loh via Facebook:
“This is a special post for friends and readers who come across misinformation about melatonin and need help in addressing that misinformation.
➡️ Melatonin production in the pineal gland at night accounts for less than 5% of total produced by the human body.
➡️ The 95%+ melatonin is produced mainly by mitochondria in every cell of the human body and is NOT regulated by light-dark circadian cycles.
➡️ Exogenously supplemented melatonin does not decrease endogenous production. Conversely, because melatonin can increase acetyl-coA production, exogenous melatonin can increase endogenous production in mitochondria.
➡️ Melatonin supplementation is critical for mitochondria because it can return pathological cells that rely on glycolysis for energy back to normal, healthy cells that produce energy via oxidative phosphorylation (OXPHOS).
* Disclaimer: These statements have not been evaluated by the Food and Drug Administration. Ascorbic acid, melatonin and any other product mentioned is not intended to diagnose, treat, cure or prevent any disease.
References:
[1] Reiter, R. J.; Sharma, R.; Rosales-Corral, S.; Manucha, W.; Chuffa, L. G. de A.; Zuccari, D. A. P. de C. Melatonin and Pathological Cell Interactions: Mitochondrial Glucose Processing in Cancer Cells. Int. J. Mol. Sci. 2021, 22 (22). https://doi.org/10.3390/ijms222212494.
[2] Reiter, R. J.; Ma, Q.; Sharma, R. Melatonin in Mitochondria: Mitigating Clear and Present Dangers. Physiology 2020, 35 (2), 86–95. https://doi.org/10.1152/physiol.00034.2019.
2 notes
·
View notes
Text
Demand for Early Treatment is Likely to Drive the Gene Expression Analysis Market
Increasing adoption of targeted molecular therapeutics and precision medicine are anticipated to fuel demand for gene expression analysis. Such kind of analysis enables scientific discoveries and supports research by explaining the functioning of genes at molecular levels. RNA Expression, Promoter Analysis, and Protein Expression & Posttranslational Modification Analysis are the common techniques of gene expression analysis.
New Analysis Technique to Promote R&D
Under RNA expression technique, DNA microarrays and PCR analysis are quite popular among researchers to study expressions of various genes and microorganisms. In addition, RNA sequencing can offer high precision analysis for broad range data. This, in turn, is likely to increase demand for RNA expression techniques over the forecast period (from 2018 to 2025).
To Request A Sample Copy Of This Report @ http://bit.ly/2LIHHuV
Many prominent companies in this field are developing new and better analysis techniques to strengthen their global presence. For instance, a team of researchers from Karolinska Institute and Max Planck Institute for Biology has found a new gene expression analysis technique to expedite disease diagnosis. The analysis reflects how dysfunction of any biological process can lead to disease. A test conducted by the team on mice has brought new opportunities for research linked to Oxidative Phosphorylation (OXPHOS) system deficiency and improved disease diagnosis.
Market Overview
The gene expression analysis market is pegged to reach USD 11.3 billion by 2025, according to a report by Grand View Research, Inc. Rising cases of chronic disorders, such as cancer, is anticipated to foster growth. Reducing costs of sequencing procedures are expected to bode well for market growth. Rising need for customized medicines is also projected to propel market growth in the coming years.
In addition, favorable government initiatives, such as health awareness activities and funding, are poised to drive market during the forecast period. Some of the prominent players operating in the market for gene expression analysis are Quest Diagnostics, Inc.; Novogene Corporation; F. Hoffmann-La Roche AG; QIAGEN, and Bio-Rad Laboratories, Inc.
For more market analysis reports, please visit https://goo.gl/TDYoXQ
1 note
·
View note
Text
#cancers, Vol. 14, Pages 4503: Crosstalk of Oxidative Phosphorylation-Related Subtypes, Establishment of a Prognostic Signature and Immune Infiltration Characteristics in Colorectal Adenocarcinoma
Oxidative phosphorylation (OXPHOS) is an emerging target in #cancer therapy. However, the prognostic signature of OXPHOS in colorectal adenocarcinoma (COAD) remains non-existent. We comprehensively investigated the expression pattern of OXPHOS-related genes (ORGs) in COAD from public databases. Based on four ORGs, an OXPHOS-related prognostic signature was established in which COAD patients were assigned different risk scores and classified into two different risk groups. It was observed that the low-risk group had a better prognosis but lower immune activities including immune cells and immune-related function in the tumor microenvironment. Combining with relevant clinical features, a nomogram for clinical application was also established. Receiver operating characteristic (ROC) and calibration curves were constructed to demonstrate the predictive ability of this risk signature. Moreover, a higher risk score was significantly positively correlated with higher tumor mutation burden (TMB) and generally higher gene expression of immune checkpoint, N6-methyladenosine (m6A) #RNA methylation regulators and mismatch repair (MMR) related proteins. The results also indicated that the high-risk group was more sensitive to immunotherapy and certain chemotherapy drugs. In conclusion, OXPHOS-related prognostic signature can be utilized to better understand the roles of ORGs and offer new perspectives for clinical prognosis and personalized treatment. https://www.mdpi.com/2072-6694/14/18/4503?utm_source=dlvr.it&utm_medium=tumblr
0 notes
Text
Biomed Grid | Calcium Dysregulation and Mitochondrial Dysfunction Form A Vicious Cycle in Parkinson’s Disease
Introduction
Neurons depend on a healthy pool of mitochondria, as they require large amounts of energy and Ca2+ buffering, yet their highly extended, complex structures and long axons [1] make maintaining and distributing mitochondria a challenging task [2]. Parkinson’s disease (PD has long been linked to mitochondrial dysfunction [3], yet why specifically dopaminergic neurons degenerate first remains to be elucidated.
Regulation of mitochondrial dynamics
Figure 1:PD-causing proteins and Ca2+ in the context of mitochondrial dynamics and PD
Mitochondrial maintenance and proper axonal distribution in neurons are possible due to the highly dynamic nature of these organelles. Almost all mitochondrial proteins are post-translationally imported into the organelle [4] and mitochondrial trafficking distributes them along the cytoskeleton across the cell [5,6]. Changes in mitochondrial morphology by fusion and fission events further adjust their functionality [7,8] and finally clearance of damaged mitochondria via mitophagy, a specific form of autophagy, contributes to maintaining a pool of healthy mitochondria [9-11]. All these processes are highly regulated to ensure proper mitochondrial func tion in response to the (sub-)cellular environment (Protein import: [4,12]; Trafficking: [9,13-16]; Fission/fusion: [17-19]; Mitophagy: [20-22]. While mitophagy has received a lot of attention as two of the main players are mutated in hereditary PD (PINK1/Parkin) [23,24], several signalling pathways disrupted in PD also alter mitochondrial trafficking (Figure 1).
The blue box lists those proteins found mutated in PD, which directly influence one or more aspects of mitochondrial dynamics and Ca2+ handling. The aspects of mitochondrial dynamics regulated are listed in the green box. The beneficial feedback cycle between Ca2+ and mitochondrial dynamics that allows adaption of mitochondrial dynamics to the cellular (sub)environment is depicted on the right. Upon misregulation of either side of the feedback loop a vicious cycle ensues that exacerbates both mitochondrial dysfunction and disrupted Ca2+ signalling in PD.
A central target of these pathways is the mitochondrial outer membrane protein Miro (also RHOT1/2) which couples the organelle to the cytoskeleton via the adaptor protein Milton (also TRAK1/2) [25-28]. Miro gets phosphorylated by the kinase PINK1 and consecutively ubiquitinated by the E3-Ligase Parkin [20-22], which leads to its degradation and mitochondrial arrest [14,29]. A parallel pathway leading to Miro degradation seems to involve LRRK2 kinase [30]. Likewise, accumulated a-synuclein also dysregulates Miro on several levels. It affects Miro subcellular distribution and limits transport of healthy mitochondria into axons [31]. Furthermore, it also prevents Miro degradation in response to mitochondrial damage [32]. Finally, cytosolic Ca2+ directly binds to the EF hands in Miro, leading to a conformational change that also arrests mitochondrial movement [13,33-35].
Calcium signalling in PD
Ca2+ is essential for cellular signalling, as it allows cells to quickly adapt and respond to the local microenvironment. The failure to control Ca2+ has devastating consequences for the cell, eventually leading to cell death [36]. Neuronal mitochondria are especially needed to buffer cytosolic Ca2+ transients elicited by neuronal activity [37]. In this way, they both prolong the signals and protect neurons from the possibly detrimental Ca2+ spikes. Hence, it is not surprising that disruption of Ca2+ homeostasis is a pathological feature of several neurodegenerative diseases including PD [38]. Several PD-causing proteins are involved in controlling Ca2+ homeostasis such as PINK1, Parkin, LRRK2 and a-synuclein [39-42]. Ca2+ mito uptake, mainly mediated by the mitochondrial calcium uniporter (MCU), is required for maintaining Ca2+ homeostasis [43,44]. Furthermore, the mitochondrial inner membrane Ca2+/H+ antiporter LETM1 is involved in Ca2+ mito uptake. PINK1 phosphorylates LETM1, thereby regulating mitochondrial Ca2+ levels [42]. Loss of PINK1 has been shown to result in disrupted mitochondrial Ca2+ transport and make neurons more vulnerable to stress [42,45].
Miro at the center of mitochondrial calcium regulation
Miro, the mitochondrial outer membrane protein, involved in mitochondrial transport has recently also been reported to be involved in Ca2+ -related processes: Regulation of Ca2+ uptake at ER mitochondria encounter structures (ERMCS) and control of mitochondrial shape in response to Ca2+.
The Ca2+ uptake into mitochondria mainly takes place at ERMCS [46]. Miro has been found to be involved in regulating Ca2+ transfer from ER to mitochondria independent of its function in mitochondrial transport and morphology. Its yeast homolog Gem1p is translocated to ERMCS regulating the number and the size of these complexes [47]. This localization is conserved in mammalian Miro [47]. A more recent study confirms the role of Miro in controlling Ca2+ mito homeostasis at ERMCS [48]. Once Miro is translocated to ERMCS, it interacts with ERMCS components modulating the Ca2+ transfer and the integrity of the complex. The localization of Miro to ERMCS is promoted by Polo kinase-mediated phosphorylation of Miro. Loss of Miro results in Ca2+ mito depletion and overexpression in Ca2+ mito overload [48] revealing the importance of the Polo/Miro signalling pathway in the regulation of Ca2+ mito uptake. Furthermore, this study shows that both PINK1 and LRRK2 are involved in this process. They function upstream of Miro and seem to control the Ca2+ mito homeostasis through Miro. In Drosophila PINK1 and LRRK2 G2019S mutants, Ca2+ mito homeostasis is dysregulated and characterized by elevated Ca2+ mito levels. Upregulated Miro levels have been found to be responsible for the dysregulation in the PD models [48]. This is in line with the fact that both PINK1 and LRRK2 are involved in pathways that result in degradation and thus downregulation of Miro [14,30]. The increased Ca2+ mito levels, present in PINK1 and LRRK2 mutants, lead to mitochondrial swelling and eventually neuronal death.
Finally, mitochondria undergo a morphology change called MiST mediated by Miro sensing Ca2+ [49]. Miro-dependent MiST is involved in mitochondrial quality control as it is required for autophagy and mitophagy. Considering the disrupted Ca2+ homeostasis in PD, MiST may be mistakenly triggered by pathologically increased cytosolic Ca2+ levels, thereby contributing to mitochondrial dysfunction.
Why are dopaminergic neurons so susceptible?
It has been shown that, with age, the amount of Ca2+ dysregulation increases [50]. Ca2+ dysregulation, however, is not ubiquitous but restricted to specific cell types. Dopaminergic neurons, the main cell type affected in PD, are autonomous pacemakers relying on L-type Ca2+ channels. It has been shown that aging is associated with increased reliance of dopaminergic neurons on these Ca2+ channels accompanied by sustained increase in cytosolic Ca2+ levels [51]. With the need to pump Ca2+ out of the cell come higher energy demands and thus an increased oxidative phosphorylation (OXPHOS) rate. The increased need to buffer Ca2+ and therefore elevat ed Ca2+ mito as well as the increased OXPHOS rate result in elevated mitochondrial oxidative stress [52,53]. Ca2+ mito overload may even directly trigger opening of the permeability transition pore (PTP) leading to the release of proapoptotic factors and even more Ca2+ into the cytosol [54]. This further exacerbates the mitochondrial phenotype due to Ca2+ dependent arrest in a vicious cycle (Figure 1) and will eventually lead to programmed cell death [55,56]. This, along with the need to feed an extensively branched axonal arbor [57], may be one of the reasons why dopaminergic neurons are particularly susceptible in PD.

Read More About this Article: https://biomedgrid.com/fulltext/volume5/calcium-dysregulation-and-mitochondrial-dysfunction-form-a-vicious-cycle-in-parkinsons-disease.000920.php
For more about: Journals on Biomedical Science :Biomed Grid | Current Issue
#biomedgrid#american journal of biomedical science & research#journals on biomedical intervention#open access journals of biomedical science
0 notes
Text
Mitochondrial Dysfunction in mtARS Disorders
Introduction
Mitochondria are indispensable organelles that facilitate cellular bioenergetics, predominantly through oxidative phosphorylation (OXPHOS). Mitochondrial aminoacyl-tRNA synthetases (mtARS) are essential for the fidelity of mitochondrial translation, catalyzing the ligation of amino acids to their cognate tRNAs. Mutations in mtARS genes precipitate a spectrum of mitochondrial disorders, culminating in dysfunctional protein synthesis and aberrant mitochondrial bioenergetics. This review delves into the molecular pathogenesis of mitochondrial dysfunction in mtARS disorders, elucidating their biochemical perturbations, clinical phenotypes, and emerging therapeutic paradigms.
Molecular Pathophysiology of mtARS Disorders
MtARS enzymes ensure translational accuracy by charging mitochondrial tRNAs with their respective amino acids, a prerequisite for mitochondrial protein biosynthesis. Pathogenic variants in mtARS genes result in defective aminoacylation, perturbing mitochondrial translation and compromising the integrity of the electron transport chain (ETC). These perturbations induce bioenergetic deficits, increased reactive oxygen species (ROS) production, and secondary mitochondrial stress responses, leading to cellular demise.
Genetic Etiology of mtARS Mutations
Dysfunctional mtARS genes such as DARS2, AARS2, RARS2, and YARS2 have been implicated in autosomal recessive mitochondrial disorders. These mutations exhibit tissue-specific phenotypic heterogeneity, with neurological, muscular, and systemic manifestations. For instance, DARS2 mutations drive leukoencephalopathy with brainstem and spinal cord involvement, whereas AARS2 defects result in a constellation of neurodegenerative and ovarian pathologies.
Biochemical and Cellular Consequences
Dysfunctional mtARS enzymes manifest in multifaceted mitochondrial deficits, including impaired translation, defective OXPHOS, and dysregulated mitochondrial proteostasis.
Disruption of Mitochondrial Translation
Impaired aminoacylation abrogates the synthesis of mitochondrially encoded proteins, undermining the assembly of ETC complexes. This translational arrest culminates in defective ATP synthesis and precipitates a systemic energy deficit.
Electron Transport Chain Dysfunction and Bioenergetic Failure
Pathogenic mtARS mutations lead to OXPHOS inefficiencies, reducing mitochondrial membrane potential (Δψm) and ATP output. Perturbed electron flux exacerbates ROS accumulation, instigating oxidative damage and apoptotic cascades.
Mitochondrial Unfolded Protein Response (UPRmt) Activation
Cellular compensatory mechanisms, including UPRmt, are upregulated in response to mitochondrial translation failure. UPRmt mitigates proteotoxic stress via chaperone-mediated protein refolding and degradation pathways. However, chronic UPRmt activation fosters maladaptive stress responses, contributing to progressive cellular degeneration.
Clinical Manifestations
mtARS disorders exhibit phenotypic variability, spanning from mild neuromuscular impairment to severe multisystemic involvement. The pathophysiological hallmark includes disrupted neurological, muscular, and cardiac function.
Neurological Dysfunction
Neurodegeneration is a predominant feature of mtARS disorders, manifesting as ataxia, seizures, intellectual disability, and progressive leukoencephalopathy. Magnetic resonance imaging (MRI) frequently reveals white matter abnormalities, indicative of compromised oligodendrocyte function.
Myopathy and Metabolic Dysregulation
Muscle tissue, with its high ATP demand, is particularly susceptible to mitochondrial dysfunction. Clinical hallmarks include hypotonia, muscle weakness, and exercise intolerance, often concomitant with metabolic anomalies such as lactic acidosis and elevated pyruvate-to-lactate ratios.
Cardiomyopathy and Mitochondrial Energetics
Hypertrophic cardiomyopathy has been observed in YARS2-associated mitochondrial disorders, wherein compromised ATP synthesis in cardiomyocytes disrupts contractile function and electrophysiological stability.
Diagnostic and Functional Evaluation
A combination of genomic, biochemical, and imaging modalities facilitates the diagnosis of mtARS disorders.
Genomic and Transcriptomic Analysis
Whole-exome sequencing (WES) and whole-genome sequencing (WGS) are pivotal for identifying pathogenic mtARS variants. Transcriptomic profiling elucidates perturbations in mitochondrial gene expression networks, further refining diagnostic accuracy.
Functional Mitochondrial Assays
Biochemical assays, including high-resolution respirometry, ATP quantification, and ETC enzymatic profiling, provide insights into mitochondrial bioenergetics. Patient-derived fibroblasts and induced pluripotent stem cells (iPSCs) serve as valuable models for functional interrogation.
Neuroimaging and Biomarker Identification
Advanced imaging modalities such as MR spectroscopy (MRS) detect metabolic derangements, including lactate accumulation in affected brain regions. Circulating mitochondrial-derived peptides and metabolomic signatures are emerging as potential diagnostic biomarkers.
Emerging Therapeutic Strategies
Despite the absence of curative therapies, multiple avenues are under investigation to ameliorate mitochondrial dysfunction in mtARS disorders.
Mitochondria-Directed Antioxidants
Therapeutic compounds such as MitoQ, idebenone, and edaravone aim to attenuate oxidative stress and preserve mitochondrial integrity.
Genetic and RNA-Based Interventions
Gene therapy strategies utilizing adeno-associated virus (AAV)-mediated delivery and CRISPR-based genome editing are being explored for genetic correction of mtARS mutations. Additionally, RNA-based approaches, including antisense oligonucleotides (ASOs) and mRNA replacement therapy, hold promise in restoring mtARS functionality.
Metabolic Modulation and Supportive Therapies
Ketogenic diets, NAD+ precursors (e.g., nicotinamide riboside), and mitochondrial biogenesis activators (e.g., PGC-1α modulators) are under investigation to enhance cellular energy metabolism. Supportive interventions, including physical therapy and neuromuscular rehabilitation, remain integral to patient management.
Conclusion and Future Directions
Mitochondrial dysfunction in mtARS disorders arises from defective mitochondrial translation, OXPHOS perturbation, and maladaptive stress responses. Advances in genomic medicine, mitochondrial therapeutics, and precision medicine approaches are poised to transform the diagnostic and therapeutic landscape. Continued research into mtARS pathobiology, coupled with translational innovations, will be instrumental in developing targeted interventions for affected individuals.

#Mitochondrial dysfunction#Aminoacyl-tRNA synthetases (mtARS)#Oxidative phosphorylation (OXPHOS)#Electron transport chain (ETC)#Reactive oxygen species (ROS)#Mitochondrial translation#Mitochondrial unfolded protein response (UPRmt)#Bioenergetic failure#Neurodegeneration#Leukoencephalopathy#Hypertrophic cardiomyopathy#Myopathy#Whole-exome sequencing (WES)#Whole-genome sequencing (WGS)#ATP synthesis#Gene therapy#CRISPR-based genome editing#RNA-based interventions#Metabolomic biomarkers#Mitochondrial biogenesis
0 notes
Text
Filip Janku, MD @FilipJankuMD @MDAndersonNews #ASCO20 Phase I study of IM156, a novel potent biguanide oxidative phosphorylation (OXPHOS) inhibitor, in patients with advanced solid tumors
http://dlvr.it/Rgl9dd
0 notes
Text
Aluminum toxicity may induce dyslipidemia and mitochondrial dysfunction in the liver through promotion of anaerobic metabolism, L-carnitine downregulation, and more.
PMID: Exp Cell Res. 2011 Oct 1 ;317(16):2231-8. Epub 2011 Jul 20. PMID: 21787768 Abstract Title: Hepatic response to aluminum toxicity: dyslipidemia and liver diseases. Abstract: Aluminum (Al) is a metal toxin that has been implicated in the etiology of a number of diseases including Alzheimer's, Parkinson's, dialysis encephalopathy, and osteomalacia. Al has been shown to exert its effects by disrupting lipid membrane fluidity, perturbing iron (Fe), magnesium, and calcium homeostasis, and causing oxidative stress. However, the exact molecular targets of aluminum's toxicity have remained elusive. In the present review, we describe how the use of a systems biology approach in cultured hepatoblastoma cells (HepG2) allowed the identification of the molecular targets of Al toxicity. Mitochondrial metabolism is the main site of the toxicological action of Al. Fe-dependent and redox sensitive enzymes in the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) are dramatically decreased by Al exposure. In an effort to compensate for diminished mitochondrial function, Al-treated cells stabilize hypoxia inducible factor-1α (HIF-1α) to increase ATP production by glycolysis. Additionally, Al toxicity leads to an increase in intracellular lipid accumulation due to enhanced lipogenesis and a decrease in the β-oxidation of fatty acids. Central to these effects is the alteration of α-ketoglutarate (KG) homeostasis. InAl-exposed cells, KG is preferentially used to quench ROS leading to succinate accumulation and HIF-1α stabilization. Moreover, the channeling of KG to combat oxidative stress leads to a reduction of l-carnitine biosynthesis and a concomitant decrease in fatty acid oxidation. The fluidity and interaction of these metabolic modules and the implications of these findings in liver-related disorders are discussed herein.
read more
0 notes