
Statistics
We looked inside some of the posts by catscoffeecancer-blog and here's what we found interesting.
Average Info
Notes Per Post
0
Likes Per Post
0
Reblog Per Post
0
Reply Per Post
0
Number of Posts By Type
Text
1
Last Seen Tumblr Blogs





Fun Fact
Tumblr is used by 21% of adults online aged 18-29 years.
Text
Why “I have cancer” doesn’t do you, or your cancer, justice.
“As we feared, it’s Cancer.”
A cancer diagnosis is often one of the things that a person fears most. After all, I’m 22 and, as I like to believe, relatively healthy but I can’t even rule it out. Anyone can be affected, regardless of race or riches. Along with a significant amount of time in hospitals, a cancer diagnosis is often accompanied by a significant amount of drugs and fear, of which fear may be more debilitating than the disease itself. Most often, these fears are anchored in the unpredictable nature of cancer itself. Cancers have this inexplicable way of changing when we don’t expect them to: maybe by responding to therapies at first then developing resistance and growing again, or maybe by coming back unexpectedly in a person who thought they had been cured for years. Nevertheless, we consistently refer to the disease as just “cancer”, or “lung/breast/colon/etc cancer”, at best. We say things like “my tumour has shrunk” or my cancer has gone away”, even though referring to cancer as one single entity is greatly oversimplifying the problem. In no way does this article mean minimize or trivialize this feeling; the cancer road is daunting, but not without hope. From a scientific perspective, I aim only to explain why your cancer can’t simply die already; rather, explain how treating your cancer as one disease is flat-out wrong. The differences between individual cells in a tumour and the concept of tumour evolution are thought to be crucial factors in the dangerous and unpredictable nature of the disease. Here, we discuss how and why tumour cells can adapt and successfully survive in a human body that just wants the cancer to die already.
Evolution: Bird beaks, and why your cancer won’t just die already.
To understand how tumours change, it is crucial to understand the concept of evolution. Specifically, how selective pressures can lead to the development of a certain trait. Charles Darwin became famous for his illustrations of this concept through rapidly-observed evolution of finches on the Galapagos Islands. These finches, while originating from the same population, rapidly developed different beak sizes so that they could best survive on their isolated islands, each with its own unique food source. Evolution allows organisms to develop in to a specific niche and succeed and thrive in their environment. While the finches developed unique beaks, the trait that changes most varies species-to-species depending on what gives them the greatest survival advantage. Cancer cells require different traits to succeed and take over our bodies: moving faster, not dying, and replicating faster, to name a few.
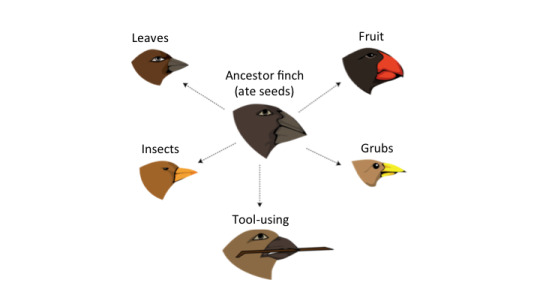
Figure 1. Darwin’s finches evolved to have different beaks depending on their food source.
Mutations can cause cancer.
So how do these characteristics come about? There has to be a reason that these cells develop the traits necessary to survive in a human body that is trying to kill them. To explain this, we have to go back to the basic where/why: DNA and carcinogens, specifically.
Carcinogens, we’ve all heard of them. But what does that really mean? How does one substance cause cancer? Not all carcinogens affect DNA directly, and they can cause cancer by other methods such as inducing inflammation (see: Hallmarks of Cancer). However, for the purposes of understanding how different tumours can be, all we really need to know is that most carcinogens act by causing damage to DNA, the string of nucleotides (A’s, G’s, C’s, and T’s) that make up the genes that code for proteins that make up the different traits in an organism (or cancer cell, in our case).
The one crucial point to remember is that by the time a tumour is detected, it is made up of millions of individual cells. Each cell has a copy of the DNA from the original cell, but it may have picked up some mistakes when the DNA was replicated along the way. As cells divide (Figure 2), they make a copy of the DNA so that it can be split evenly into two identical cells. Most of the time this works near-perfectly, in that in the 3 billion A’s, G’s, C’s, and T’s in one strand of the human genome, only 0.000001% will be wrongly copied and left un-fixed. However, not all mutations are created equal: a mutation at just any spot in the cell’s DNA might not make a difference. For a mutation to change a trait, it has to (for the most part) occur in a region that codes for a protein (approximately 1% of the DNA does this). Mutations are like throwing a dart at a wall of tiny nucleotides, it could hit anywhere, but on the off chance it hits that 1% region, there is a limited chance that it causes a change in the protein. Even then, few of these changes are associated with a fitness advantage (which from Darwin, we learned are more likely to survive because of evolution). However, if this function is affected, a mutation can cause a change in a trait. In the case of a cancer cell, this may cause the cell to replicate and grow faster, move throughout the body, or just not die, to name a few of the characteristics that help a cancer cell survive in a human body that is trying to fight off the disease.
The most widely accepted theory relies on one fact: that tumours arise from one single cell that contains specific DNA mutations that caused that cell to develop those cancerous behaviours (keep growing and not die; herein referred to as ‘founder’ mutations). This theory refers to cancer cells that descend from this cell as clones, as they contain these founder mutations. However, cells can pick up additional mutations along the way, making them genetically distinct (Figure 2).
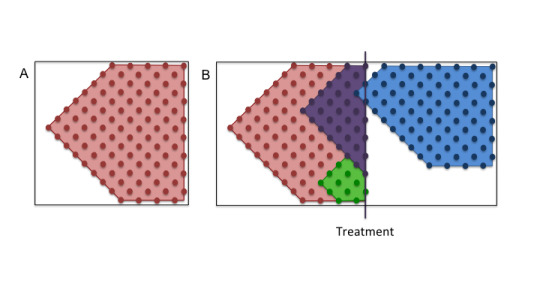
Figure 2. Cells divide and pick up new mutations along the way. B) Treatment can kill off specific cell populations, but not others.
Clonal heterogeneity: Why different clones can exist in one cancer.
The differences between tumour cells in cancer are driven by two main factors: the introduction of DNA mutations, and the existence of evolution (which allows for the selection of particular mutations that result in an advantage in the organisms ability to survive, as we discussed earlier). However, the presence of any mutation, provided it doesn’t make the cell more vulnerable, provides an advantage: heterogeneity in general is a trait that helps tumours diversify, and thus increases the cell populations ability to adapt to a wider range of stresses. This results in cancers that are comprised of millions of cells with variations in their DNA (see Figure 3). So basically, if a mutation is induced, that cell is likely to succeed in the tumour cell population. Unless, of course, it makes the tumour cell more vulnerable, which is bad for it, but great for you.
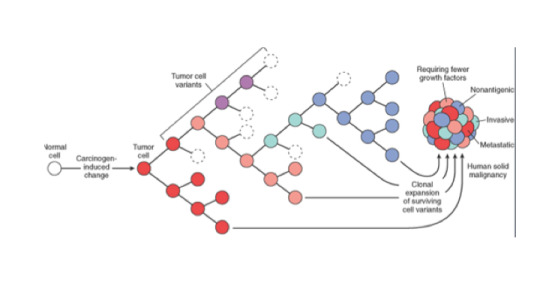
Figure 3. Cells with different DNA make up a tumour.
Tumour heterogeneity and personalized medicine: Customizing cancer treatment.
The most famous genius once said: “Everything should be made as simple as possible, but no simpler” (if you didn’t immediately think of Einstein, Newton would do. But for the record, it was Einstein). While current cancer treatments have the ability to kill cancer cells, the fatal flaw in most cancer treatments is that they greatly underestimate the complexity and variability in each tumour. This heterogeneity is pervasive: in multiple samples taken from different regions of the same tumour in one person, it was found that 63-69% of DNA mutations found in a single sample were not uniformly detected in the other samples from the same tumour. This tells us it is not only crucial that therapy be customized for each patient, but also specifically customized to each component of a given tumour. However, it is not always possible to capture all the regions of the tumour, because getting a sample of what the tumour looks like often involves surgery, and even then, many tumours aren’t in an accessible area of the human body. These unavoidable technical problems with sampling prevent doctors from knowing all they can about the tumour. Thus, the treatment can only focus on the areas of the tumour that we know about, which may or may not be representative of the entire tumour.
How to kill a cancer: How can this variation in tumours be drugged?
Ideally, a cancer drug would target every cancer cell specifically, without affecting normal cells. These specific drugs can select only cells that contain specific mutation; ideally, one that persists on a large portion of the cells and cause the size of the tumour to regress. Ideally, the target of this therapy regime would be a founder mutation, or a mutation that is present in every cell. However, these mutations are not always clear to the researcher or physician, as it is hard to know everything about the tumour without taking the whole tumour out of the patient. Thus, treatment regimes settle on mutations that already have drugs developed, relying on combination therapies if necessary.
If one drug doesn’t kill all the cells, why not use multiple drugs at once? Unfortunately, easier said than done. Combination therapies are challenging because the effectiveness relies on the mutations in the tumour, and are thus extremely patient-specific. Thus, the combination of effective drugs could drastically vary patient-to-patient. However, the study of combination drug efficacy on a large scale presents many challenges, as it inherently relies on patient specificity, resulting in a vicious cycle. Additionally, before treatments such as these are introduced on a large scale, these combinations must be well-studied to determine if they produce any undesired effects when they are delivered to the patient at the same time to avoid unwanted patient toxicities.
Herein lies the key to why most clinical trials fail: for the most part, drugs aren’t tested only on people with cancers that could be affected by the drug (based on the DNA in the cancer cells themselves). In a perfect world, the assessment of drug efficacy would only include patients that have tumours with a genetic background that can be targeted by the drug. Due primarily to technical limitations, clinical trials do not provide information about how specific clones in a tumour perform in response to each selected drug. It is surgically impossible to sample all the sites of metastasis in a person. A lack of response by one specific subclone in the body may result in an observable decrease in the size of the tumour, if most cells are affected, but may leave another subclone unaffected and free to grow and take over the tumour. This process can result in a disease that appears to not respond to the treatment it is being administered (Figure 3). Therefore, to truly understand disease progression, is crucial to understand how the heterogeneity of a tumour changes over time in response to a specific drug.
Now you know?
Once you know, it is impossible to look at cancer as one disease. It is comprised of cells that may all respond differently to anything you throw at it, and by calling it only ‘cancer’, we are greatly oversimplifying the problem. Knowing that cancers can’t be treated as one disease allows the patient to better understand the behaviour of their disease, and hopefully, understand that when a cancer ‘comes back’, it may not look the same as the one you started with. I don’t say this to be dismal; in fact, this could mean that you are more likely to respond to the second drug they give you, and have an entirely new chance for the therapy to work, because really, you might not be dealing with the same disease at all. Everything about cancer is complicated; from the DNA changes that make tumours so different, to going through treatment. The more you know, the better you can be at facing the disease head on, and giving the fight your all.
References
McGranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell. 2017 Feb 9;168(4):613-628. PMID: 28187284.
Venkatesan S, Swanton C. Tumor Evolutionary Principles: How Intratumor Heterogeneity Influences Cancer Treatment and Outcome. Am Soc Clin Oncol Educ Book. 2016;35:e141-9. PMID: 27249716.
Alizadeh AA, Aranda V, Bardelli A, Blanpain C, Bock C, Borowski C, Caldas C, Califano A, Doherty M, Elsner M, Esteller M, Fitzgerald R, Korbel JO, Lichter P, Mason CE, Navin N, Pe'er D, Polyak K, Roberts CW, Siu L, Snyder A, Stower H, Swanton C, Verhaak RG, Zenklusen JC, Zuber J, Zucman-Rossi J. Toward understanding and exploiting tumor heterogeneity. Nat Med. 2015 Aug;21(8):846-53. PMID: 26248267
Aparicio S, Caldas C. The implications of clonal genome evolution for cancer medicine. N Engl J Med. 2013 Feb 28;368(9):842-51. PMID: 23445095.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646-74. PMID: 21376230.
0 notes