
Don't wanna be here? Send us removal request.
Statistics
We looked inside some of the posts by labdiary and here's what we found interesting.
Average Info
Notes Per Post
0
Likes Per Post
0
Reblog Per Post
0
Reply Per Post
0
Time Between Posts
6 months
Number of Posts By Type
Text
13
Quote
1
Link
1
Photo
1
Video
1
Last Seen Tumblr Blogs





Fun Fact
In February 2021, Tumblr had 518.6 million blog accounts.
Text
Media selection
https://www.americanpharmaceuticalreview.com/Featured-Articles/559489-CHO-Media-Development-for-Therapeutic-Protein-Production/ Product Quality Considerations
Although cell growth and productivity have been the focus of media development research, particular attention should be paid to product quality throughout the optimization process since it is closely associated with efficacy, potency, and safety of therapeutic proteins. Media components have been reported to impact many product quality attributes, including glycosylation, charge variants, size variants, and sequence variants.30 The effects of selected media components on these product quality attributes are exemplified in some case studies as follows. Copper has been widely reported to increase basic charge variants and affect mAb aggregation and fragmentation.31-33Manganese is a well-known factor to modulate glycosylation pattern, such as high mannose species, galactosylation, sialylation, and glycation.30 Cystine, asparagine, and glutamine have been found to increase sialylation while cystine was also described to decrease acidic charge variant and low molecular weight species.30,34,35 In addition to their positive effect on titer, hydrocortisone and Long®R3 IGF-1 were shown to increase the sialylation level of an Fc-fusion protein significantly.16,18 Non-nutritional components, such as acetylated N-acetylmannosamine and glycerol, were also indicated to improve protein sialylation.36,37 In terms of amino acid misincorporation, an efficient preventative step is to provide sufficient amino acids to keep culture from exhausting those nutrients.38
0 notes
Quote
Finite vs Continuous Cell Line Normal cells usually divide only a limited number of times before losing their ability to proliferate, which is a genetically determined event known as senescence; these cell lines are known as finite. However, some cell lines become immortal through a process called transformation, which can occur spontaneously or can be chemically or virally induced. When a finite cell line undergoes transformation and acquires the ability to divide indefinitely, it becomes a continuous cell line.
https://www.thermofisher.com/us/en/home/references/gibco-cell-culture-basics/introduction-to-cell-culture.html
0 notes
Link
From the Cultural Revolution to the Gene Therapy RevolutionMay 4, 2020
Guangping Gao, PhD, is professor and director of the Horae Gene Therapy Center at the University of Massachusetts Medical School in Worcester, MA. Over the course of three decades, Gao has made profound contributions in the area of adeno-associated virus research, initially working with James M. Wilson, MD, PhD, director of the gene therapy program at the University of Pennsylvania. Gao has received multiple honors in recognition of his service, expertise, and dedication. For example, he was named president (2019–2020) of the American Society of Gene and Cell Therapy.
Gao has published 250+ research papers, six book chapters, and four edited books, and has fulfilled editorial responsibilities for several gene therapy and virology journals, including the Human Gene Therapy, a journal that Gao currently serves as deputy editor-in-chief. Gao recently spoke to Kevin Davies, PhD, executive editor of Human Gene Therapy, about his remarkable life journey and hopes for the future of gene therapy. (The interview originally appeared in Human Gene Therapy, Vol. 31, Nos. 3 and 4, published by Mary Ann Liebert. Kevin Davies, PhD, executive editor of Human Gene Therapy, conducted the interview.)
We will get to your preeminent research and leadership in the gene therapy field, but let’s start at the beginning.
Gao: I grew up in China during the Cultural Revolution. Around 1975, I was compelled to leave my studies and go to the countryside to receive additional “education” from farmers and peasants. My dream about new medicine really starts there. I interacted with farm laborers on a daily basis, and I saw many of them suffer from various diseases and painful conditions.
I was trying my best to use acupuncture and traditional medicine to help them, but I wished I could have some “magic medicine” to make a more substantial impact, particularly for the elderly and people with cancer.
In 1978, I was one of the first generation of students to enter college after the Cultural Revolution. I was admitted to a medical university in Chengdu, Sichuan. I worked on drug development and medicinal chemistry. In 1988, I graduated from the university and got an opportunity to come to the United States, sponsored by the World Health Organization (WHO). I was looking for opportunities to develop the next generation of medicines that I had dreamed about back on the collective farm.
I started my PhD at Miami Children’s Hospital and Florida International University with my mentor, Reuben Matalon, a pediatrician and medical geneticist. He was a prominent researcher on rare diseases such as Tay-Sachs, Hurler, and Gaucher. His major contribution as a geneticist was the discovery of the biochemical defect in an inherited leukodystrophy called Canavan disease.
I remember it well—I published that paper in the early days of Nature Genetics!
Gao: Yes, thank you! I joined his lab in 1989. My assignment was to isolate the genes and the mutations responsible for Canavan disease. Working with my lab mentor, Rajinder Kaul, I discovered the gene and mutations for Canavan disease and published my thesis work in Nature Genetics in 1993.1
After that, I asked myself, what’s my next step? Because we saw many Canavan patients at these centers, we knew exactly what was going wrong with those kids. We had to figure out a way to fix it. In 1993, I decided to look for the next generation of medicine, specifically at the opportunities in gene therapy for genetic disorders. Finally, Jim Wilson accepted me as a postdoctoral fellow at the University of Pennsylvania’s Institute for Human Gene Therapy.
The first task Jim gave me was to create new generations of adenovirus. At that time, adenovirus vector was much hyped because it has a high transduction efficiency. Because we knew adaptive immunity/immunotoxicity is a major issue for adenovirus, we decided to cripple the virus further to make it more replication defective. This might prolong transduction efficiency and stability in tissues.
I spent about two years there, first making a cell line to complement the crippled virus. Then we used that cell line to create the further-crippled virus. (You need to transcomplement its growth with E1 and E4.) They called this third-generation virus at the time. We demonstrated that, yes, virus can reduce liver toxicity in mice and immunotoxicity and prolong expression substantially.
When I published that work in 1996,2 I said to Jim, “I’d like to move on and start my career in industry because I have two kids to raise.” I was 38 at the time. He said, “No! Why leave? I’m going to give you a job.” He told me they were trying to apply the next-generation adenovirus vector for clinical trials. There was a lab called the Human Applications Lab, a GMP facility at Pennsylvania Hospital where scientists were trying to grow the virus for multiple clinical trials, but they could not grow it well.
My career in gene therapy started from there. I spent about two years making the virus work. In the first two weeks, I was able to generate high quantities of virus. Jim was in his office, talking to a reporter from the Philadelphia Inquirer. I told Jim, “I got the virus, and they are 1013 or 1014.” Jim said to the reporter, “Now we can even swim in this gene therapy vector!”
By that time, we were doing several clinical trials in cystic fibrosis, ornithine transcarbamylase (OTC), mesothelioma, and others. By early 1998, we wanted to look for new viruses, the next generation of gene delivery vehicles. I started working with AAV prototypes such as AAV-2, AAV-1, and AAV-5. Those were the first serotypes to attract a lot of interest and development.
Who first identified AAV? Was it discovered serendipitously?
Gao: Yes, it was discovered in 1965 from some adenovirus preps. They called it adeno-associated virus (AAV) because when they purified the adenovirus and looked at it under a microscope, it was a very small virus in the company of the much larger adenovirus.3 I think Arun Srivastava and others sequenced AAV. Nick Muzyczka, Jude Samulski, Barrie Carter, and others started vectorizing—demonstrating you can create a vector in transduced cells very easily. Many groups then demonstrated that AAV can transduce animals in vivo. The difference is that adenovirus only sustains for a maximum of two to four weeks. But AAV—at that time, primarily AAV-2—can sustain for hundreds of days.
My first task with Jim was to figure out how to produce a scalable manufacturing process. I started making cell lines, creating adeno-AAV hybrids. I published a paper in 1998.4 We converted a transfection-infection system into a total infection system that generates tons of AAV. Working with my colleague Guang Qu, we developed a column purification system using heparin-binding columns in early 2000.
Then on September 17, 1999, this tragic event with adenovirus OTC gene therapy happened, and we lost 19-year-old Jesse Gelsinger. For the entire field, it was a drop from a peak to a deep valley. We experienced 10 years of dark ages for gene therapy. I continued my AAV work. We started the first AAV-2 limb-girdle dystrophy clinical trial with Jerry Mendell (Nationwide Children’s Hospital, Columbus, OH) and colleagues at Penn such as Hansel Stedman and Lee Sweeney. We started the trial using the vector produced with my manufacturing methods under GMP conditions.
After the Gelsinger tragedy, was there added urgency and commitment to establish AAV as an alternative vector?
Gao: Absolutely. We started working with adenovirus, based on the discovery by Yiping Yang (formerly at Duke, now at Ohio State). He discovered immunotoxicity of adenovirus. My job was to reduce that adaptive immunity to adenovirus. But we overlooked this innate immunity, this cytokine storm, which killed Gelsinger.
I had initially started with AAV-2, but we did not really think about AAV-1 and AAV-5, or about discovering new AAVs, until Gelsinger. Then we realized, when you compare the two vectors, adeno is much more efficient. But for immunotoxicity, AAV is much, much better than adeno. Jim and I thought, if we can find a virus as efficient as adeno but without immunotoxicity, that should be the future of gene therapy. Gelsinger was an additional driving force for me to discover new AAVs.
I started work in 2001, and soon we discovered a library of new AAVs in nonhuman primates. We published our first paper in 2002.5 That paper became the hottest paper in the field and gave us new hope to work on the next generation of gene therapy vectors.
How did that discovery come about?
Gao: Back in the winter of 2001, after we found some virus sequences, I presented the PCR data to Jim Wilson at a lab meeting. I could tell his mind was spinning:“Is this real or not?” After the meeting, he said, “Guangping, I think you stepped on a goldmine.”
I started with nonhuman primates. We found that we can detect AAV in any animal. You never run into anyone with absolutely no AAV. It is in any tissue. In any PCR reaction, I always found multiple AAVs. That tells you how diverse [it is], how rapidly AAV is evolving. Then we published our second paper about nonhuman primate viruses, demonstrating AAV evolution.6
At what point did you expand or focus the search for new AAVs in humans?
Gao: You can find AAV everywhere. You can find a different AAV in the same samples. That’s why AAV is amazing to me! As the initial discovery was based on nonhuman primates, I asked Jim in late 2002, “Should we move into human tissues?” He agreed. We discovered AAV-9, which is the first “super virus” for gene therapy from humans, in January 2003.7 Our objective was to develop AAV to be as potent, as efficient, as adenovirus for transduction. But we wanted them to have much less immunogenicity. I think we accomplished that (Figure 1).8
We did not go through the traditional viral isolate characterization. We focused on PCR amplification of the capsid because we realized biology is only determined by the capsid. We didn’t need anything else. We designed PCR primers in the conserved region and amplified through hypervariable regions, generating a new virus capsid with new biology.
When did you move to the University of Massachusetts?
Gao: I moved in 2008. At the time, under the Life Sciences Initiative, then-governor Deval Patrick gave $1 billion to promote biomedicine in the state. Our dean, Terry Flotte, and the chancellor, Michael Collins, wanted to take the momentum to set up three centers in gene therapy, stem cells, and RNA interference. They recruited me from Penn to UMass to set up the gene therapy center.
I continued my AAV discovery, and collaborating with Terry and others—including researchers at the New Iberia (Louisiana) Research Center, a non-human primate facility—we were able to get some primate tissues and start to look for AAV from chimpanzees. We discovered hundreds of AAVs similar to AAV-1, AAV-6, AAV-4, AAV-3, AAV-5, and even AAV-9, which I discovered from humans. I did not know other primates also have AAV-9.
How would you describe the repertoire of AAV vectors? To what degree can researchers adapt these vectors?
Gao: We have now isolated new AAVs from 850 human surgical tissue samples. And we have about 1100 new AAVs. We found large amounts of AAV-2, AAV-3, and AAV-8 in human tissues. My AAV-8 was initially isolated from monkey lymph nodes, but now we see it everywhere in humans. If you talk about the natural reservoir of AAV, I think there is still a lot there.
Of course, now the field has moved to new directions. In addition to a natural reservoir, scientists have started doing directed evolution, rational design, and machine learning. They will complement our original discovery. In the AAV field now, in the clinic, I’d say 98–99% is still the natural AAV as a gene therapy platform, but there are many other AAVs in development by those other methods.
What are the remaining hurdles? Is manufacturing still a challenge?
Gao: If we want to develop clinical AAV gene therapy and commercialize the drugs, we have to overcome four barriers:
Manufacturing. Currently, if you want to use a gene therapy for eyes, for localized delivery to the brain, you don’t need much. Current technology is good enough. But if you want to do things like Duchenne muscular dystrophy or cross the blood-brain barrier, it may require up to 1016 viruses for each patient. In commercial terms, the current maximum scale is probably 1018. But if you are going to use gene therapy and commercialize the drug, usually you need to be on a scale of 1020. We are at least one or two logs away. Generating large quantities of highly potent virus is the number-one barrier we face in the field. This contributes to a major portion of the high cost of gene therapy.
Immunotoxicity. As we are giving AAV at much higher doses, preexisting immunity, innate immunity, and adaptive immunity to capsids and transgenes will become an issue. Some immunotoxicity with high-dose injections is starting to show up. We have to manage this.
Choice. People ask me, “Which AAV do you recommend if I want to target the brain?” That’s a hard question because my understanding, based on natural AAV, is you can either have an efficient or inefficient AAV. There is really a lack of a true tissue tropism, a true cell or tissue specificity. It doesn’t matter how you create a new AAV, that is the area we have to fight for. Eventually we will get there. We’ll make a designer AAV for a certain disease and certain targeted tissue.
Expression. When we do gene therapy, we typically think the more expression, the better. Soon, we will realize that sustained expression at a high, superphysiologic levels may not be good. Particularly with some haploinsufficient diseases, you may run into problems.
References 1. Kaul R, Gao GP, Balamurugan K, Matalon R. Cloning of the human aspartoacylase cDNA and a common missense mutation in Canavan disease. Nat. Genet. 1993; 5: 118–123. 2. Gao GP, Yang Y, Wilson JM. Biology of adenovirus vectors with E1 and E4 deletions for liver-directed gene therapy. J. Virol. 1996; 70: 8934–8943. 3. Hastie E, Samulski RJ. Adeno-associated virus at 50: A golden anniversary of discovery, research, and gene therapy success—A personal perspective. Hum. Gene Ther. 2015; 26: 257–265. 4. Gao GP, Qu G, Faust LZ, et al. High-titer adeno-associated viral vectors from a Rep/Cap cell line and hybrid shuttle virus. Hum. Gene Ther. 1998; 9(16): 2353–2362. 5. Gao GP, Alvira MR, Wang L, et al. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 2002; 99: 11854–11859. 6. Gao G, Alvira MR, Somanathan S, et al. Adeno-associated viruses undergo substantial evolution in primates during natural infections. Proc. Natl. Acad. Sci. USA 2003; 100: 6081–6086. 7. Gao G, Vandenberghe LH, Alvira MR, et al. Clades of adeno-associated viruses are widely disseminated in human tissues. J. Virol. 2004; 78: 6381–6388. 8. Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Disc. 2019; 18: 358–378.
0 notes
Text
Databases in immunology
Came across there important databases
IMGT
Hcdm
IPD IMGT/HLA
TCGA The Cancer Genome Atlas
GTEx Genotype Tissue expression
0 notes
Text
Immunotherapy
For the longest time having read primarily about T-cell based immunotherapies, I found it easy to forget that T-cell based therapies are just one of many approaches in the broader category that are called immunotherapies. The human immune system is complex, and multiple players in that system can potentially be recruited. Other players in the system include..
- T-cells
- Bcells
- Tumor infiltrating Lymohocytes
- Natural killer cells
- Macrophages
0 notes
Text
Isothermal Calorimetry
ITC is one of many biophysical characterization techniques that can give dissociation constants of a binding event. Additionally ITC can provide information on the entropy, enthalpy and consequently the free energy directly from the experiment. At the core of the technique is the measurement of heat added or removed (as measured by heat addition to reservoir - there is no cooling in ITC) in response to mixing of two components. The measured quantity is the power in microcalories required to maintain the constant temperature difference between the cell and the reference. Air bubbles are a major cause of erratic results.
When doing a water-water titration experiment, values close to 15DP and a stable curve indicate successful experiment.
This paper connects ITC experiments and computations; http://www.ncbi.nlm.nih.gov/pubmed/11785756
0 notes
Text
Antibody-drug Conjugates
The drug Herceptin [1] made by Genentech was approved by FDA in 1998 for treating breast cancer. I first came across the drug while studying the solutions (sugar-water mixtures) used to increase its shelf life. While the drug is quite effective in killing cancer cells, it does have significant side effects. The mechanism of the drug's effect on cancer cells is a good read in itself [2]. Very briefly, the protein HER2 is necessary for cell growth and division. It is over-expressed in cancer cell. Herceptin selectively binds to HER2 and impedes its action thereby preventing the cancer cell from dividing and proliferating.
Antibody proteins have long been used to selectively bind to specific targets. In cancer therapy these targets could be proteins or parts of DNA that are over-expressed or specifically found only in cancer cells. While antibodies can themselves be drug candidates, their ability to selectively and strongly bind to their targets makes them highly attractive as carriers of potentially more effective drugs.
To improve the efficacy of Herceptin, another drug Mertansine can be given simultaneously. The new drug binds to tubulin and further disrupts the cell division process by preventing the formation of microtubules. The disruptive action of Herceptin and Mertansine can be enhanced and more targeted by covalently linking them. This is precisely what the new drug Kadcyla [3] does. The antibody here is Herceptin (itself a good drug) and the payload it carries with it is Mertansine.
Such covalently linked antibody-drug compounds are, unsurprisingly, called antibody-drug-conjugates (ADC) [4]. To date FDA has approved only three ADCs of which one has been withdrawn from the market leaving two.
1. http://en.wikipedia.org/wiki/Trastuzumab
2. www.nature.com/nature/journal/v421/n6924/full/nature01392.html
3. http://en.wikipedia.org/wiki/Trastuzumab_emtansine
4. http://en.wikipedia.org/wiki/Antibody-drug_conjugate
0 notes
Text
dNTP vs ddNTP
Back in the 70's Sanger developed an ingenious way to find the sequence of a DNA strand. His approach was to first throw in DNA polymerase and normal dNTPs (i.e. deoxy-Adenosine-Tri-Phosphate, deoxy-Guanosine-TP, deoxy-Thymidine-TP and deoxy-Cytidine-TP). dNTPs look like so...
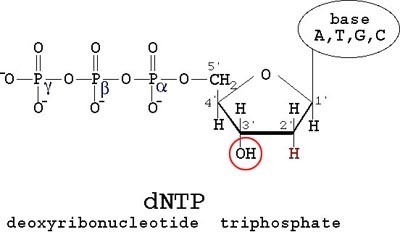
The thing about dNTPs is that they are the monomers used to generate a chain of DNA. But when a di-deoxy-NTP is thrown in the mix and is recruited by DNA polymerase, it leads to chain termination since a ddNTP has only one reaction site. A ddNTP looks like so...
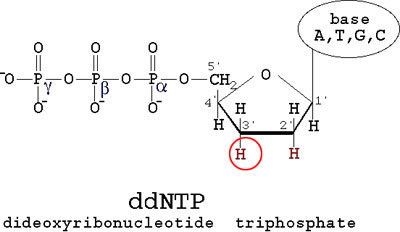
Now you can get ddATP, ddTTP, ddGTP and ddCTP. By throwing in a ddNTP of choice in to a mix of dNTP (notice the single d), you can stop the chain at you choice of A/T/G/C. This will lead to chains of various lengths terminated by A/T/G/C. When run through a electrophoresis column, you get distinct peaks corresponding to different lengths. When peaks of A/T/G/C are put together you get a nicely separated spectrum which gives you the sequence of your DNA. A sample spectrum looks like so...

Here each of the 4 colors corresponds to the 4 nucleotides. Figures below add to this discussion.
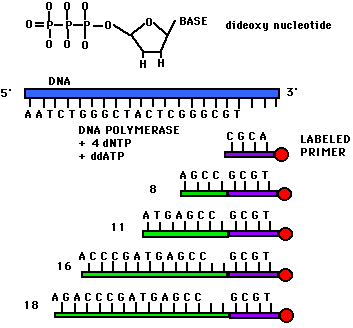
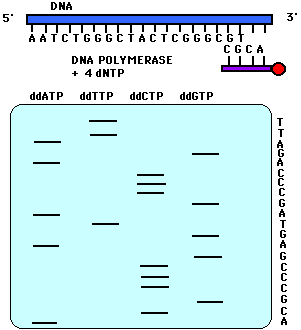
References:
Figures 1-3 are from
http://www.ocf.berkeley.edu/~edy/genome/sequencing.html
Figures 4-5 are from http://www.bio.davidson.edu/Bio111/seq.html
0 notes
Text
Story behind this gel

This polyacrylamide gel was used to verify the expression and purification of the protein L30E. The black oval on the left is the mark left by the pure protein. The black rectangle is the lane formed by a mixture of proteins of known molecular weights. With these two lanes as the benchmark let us look at the results from the flow through. What is flow through?
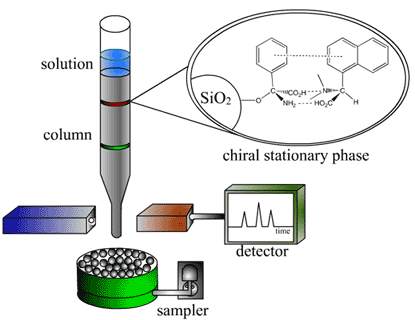
The output of a chromatography column (metal, sugar or whatever kind you use) flows at a rate of, say, 4mL/minute. Further let's say you have a volume of 80mL that contains all sort of crap along with your protein. This will take 20minutes to flow through the column. To achieve good fractionation, it is a good idea to collect the 80mL in 20 separate test tubes. A fraction collector is used for this purpose.

Now particles that do not stick to the column flow first. Say between test tubes 1-10. At this point you introduce a buffer that essentially kicks your protein (that is currently bound to the column) and it begins to show up in the fraction collector for the remaining 10 tubes (11-20). Now if your chromatography column functions properly, you should see a clean narrow band corresponding to your protein on the polyacrylamide gel. The red and blue rectangles from the first figure are from the test tubes 1-10 which is the flow through. This flow through should not have contained any amount of the protein you are trying to purify. But here you see that not only does the flow through have L30E, but the quantity is significant. If the flow through were not checked, you could have had a very low yield due to this wastage. Further the blue rectangle is interesting since it clearly shows that it is the flow through. See how it carries proteins of all possible sizes in it. This is possible only in the flow through. Moral of the story is check the full range of the fractions from the collector for traces of the protein of interest.
0 notes
Text
Protein expression, purification and characterization (NMR for now)
To perform experiments on a protein of interest, you can either buy the protein from any of the vendors or express the protein yourself. Since bacteria have a doubling time of ~30minutes, it is a good idea to use them as an agent for obtaining your protein. Now the bacteria you choose might make your protein for its own needs. But the quantity of protein generated is optimized for the bacteria's needs which in most cases is way below the amount you want to collect. So, a plasmid (circular DNA encoding the protein of interest) is introduced into the host bacteria (typically E. Coli) either by a heat-cool cycle or by electroporation.
Detour: Introducing plasmids. The term biologists use for this process is transformation, not insertion. The term insertion is used when the foreign DNA (plasmid) is inserted and made to be a part of the host DNA. This is what the HIV virus does. Moving along, how do you ensure, after introducing your plasmid, that only plasmid-carrying bacteria replicate? This is achieved by introducing antibiotic immunity into the plasmid. This way only bacteria containing the plasmid can survive a heavy dose (how much?) of the antibiotic of choice (Kanamycin, Ampicillin, find more).
The plasmids inserted usually have some form of a trigger which tells the bacteria when to read the plasmid and express the encoded protein. Plasmids are kept inactive by design where two parts (something to do with Lac) of the plasmid interact so strongly that the bacterial DNA polymerase is not allowed to bind to the plasmid DNA. But upon the introduction of a trigger compound (like IPTG), the bound regions of the plasmid let go of each other and the bacterial DNA polymerase transcribes plasmid DNA into mRNA and translates this mRNA in to the encoded protein.
Steps involved in protein expression
Grow bacteria in a rich media (50 mL LB).
Transfer this culture to minimal media containing N(15)H3Cl as the sole source of Nitrogen and C13 enriched glucose as the sole source of Carbon. The reason behind this is that you want your final protein to have enriched N and C in its backbone and as the title suggests, you want to use NMR to study the protein structure. The only problem with this media is that the bacteria has to go through numerous steps of metabolism to generate required amino acids making it a rather undesirable environment to grow in. So additional care is required to ensure bacterial reproduction in the minimal media (define additional care).
After enough bacteria (Optical density of 0.4?) has grown in the smaller volume of the minimal media (50mL), transfer this into the larger (3L) flask. Remember that the plasmid trigger (IPTG) has not yet been added. Check for OD in the larger flask and when it nears 0.4, introduce IPTG. This ensures that maximum number of viable cells are in the volume when the trigger is introduced. Leave this overnight at optimal temperature and mixing. By the next morning you should have a good amount of your protein in the flask. Subsequent steps involve separation of various components.

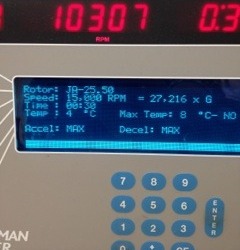
Figure: Avanti J-20 XPI Centrifuge. It is important to balance the weights of the test tubes since the centrifuge spins at 15000RPM generating about 27000 G. Any imbalance will likely damage the centrifuge beyond repair.
Spin contents of the flask at 4000rpm to isolate the cells. Drain the liquid and re-suspend the pellet in a lysis buffer. This buffer, as the name suggests, helps with cell lysis. Now cell lysis can be done in many ways(*). Mechanical lysis is one such method. Here, cells suspended in lysis buffer are pushed at high velocities (how much?) through a narrow pore. This generates enough pressure to rupture the cell membrane and lyse the cell. For high yield, re-route the output of the microfluidic chamber back into the input container so you can achieve multiple rounds of lysis. After few such cycles, collect the output and spin it in a centrifuge at 15-20,000rpm. Higher rpm is needed at this step since we want to separate insoluble (lipids) from the soluble (DNA, all proteins). The supernatant now has water-soluble compounds.


Figure: High-pressure microfluidic channel that ruptures the cell. In the second picture the output of the channel (small tube) is being reintroduced into the input tube (steel pipe).
At this stage you may want to separate DNA (why). This can be done via chemical means. PolyEthyleneImine (PEI), ethanol or Streptomycin (among others) can be used to precipitate DNA from the lysate.
Now you have water-soluble proteins in your mix. There has to be some way to distinguish your protein from other proteins. This is usually done by introducing a Histidine tag (clearly this must be encoded in the original plasmid). The rational behind having a hexa-Histidine tag is that the supernatant is run through a Nickel-coated resin (sepharose/agarose resins) which coordinates with the Histidine tag and thus only your protein binds to the column while the rest are flushed out. This separation technique is called Metal Affinity Chromatography. In a similar fashion, one can attach maltose binding protein (MBP) to the protein of interest and separate using a maltose-resin or one can attach a GST-tag to protein of interest and use GSH coated beads as described in the figure below (taken from Wikipedia).

While the Histidine tag was critical in the isolation/purification process, it could potentially alter the structure of the protein of interest. Therefore it might become important to get the protein rid of the tag. Exopeptidases are used for this purpose. Look up what Endopeptidases are used for.
Once you have only proteins suspended in solution, you might need to measure its concentration. This is achieved by;
- UV [link]
- Bradford Assay
- Lowry Assay
At this point you might want to concentrate your protein for crystallization.
Important additives at various stages:
PMSF (phenylmethylsulfonyl fluoride) is added to the cell lysis buffer since it is a protease inhibitor. You need a protease inhibitor in the mix when your cells are being lysed because you want to increase the yield of your protein. Proteases are enzymes that digest all sorts of proteins. All living cells contain proteases for a variety of reasons primary among which is to digest foreign proteins rendering immunity from potentially toxic proteins. Proteases are also used to regulate the quantity of proteins in the cell. Not all proteins are needed in high quantities at all times during a cell's life. PMSF is a rather unstable compound and has a half-life of 30 minutes in water at 25C. Therefore it is mixed in isopropanol at low temperature to keep it active during lysis.
The cell lysis buffer is an important component that will affect the yield of your protein. The buffer will vary depending on the protein of interest. Maltose Binding Protein (MBP) for example requires a pH of 7. This is achieved by using Sodium Phosphate as the buffer.
Detour:
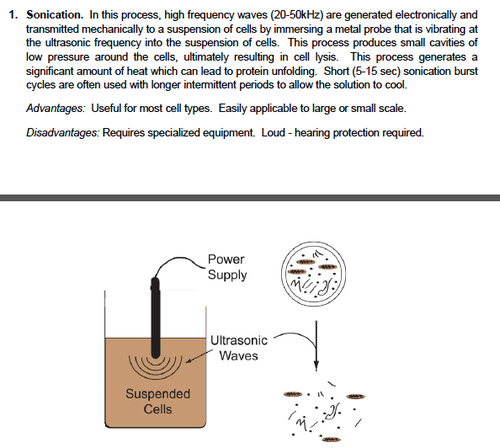
(*)Chemical lysis is a possibility. Sonication is an elegant method which splits the cell membranes and the DNA. This is useful since DNA removal is important in some cases.


Useful links:
http://proteincrystallography.org/protein-purification/
http://www.piercenet.com/method/overview-protein-assays
http://www.scribd.com/doc/183046415/Cell-lysis-protocol
http://www.scribd.com/doc/183057608/Measuring-protein-concentration-using-absorbance-at-280-nm-pdf
0 notes
Photo
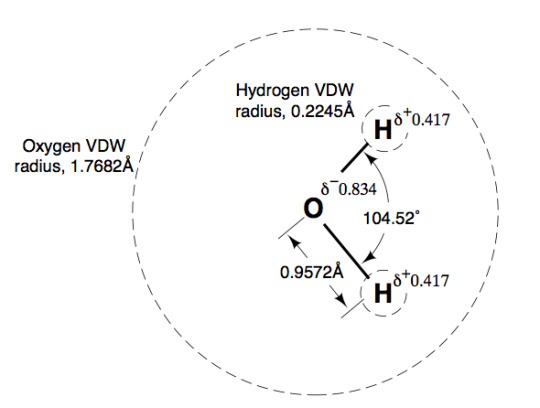
I knew the O-H bond distance and the vdw radius of Oxygen but never put the two together. Interesting to see Oxygen's vdw completely envelopes both hydrogen atoms.
0 notes
Video
youtube
Yesterday while making LB agar plates I ended up with bubbles which can be mistaken for bacterial colonies leading to considerable wastage of time. This video shows a neat trick to get rid of bubbles using a bunsen burner. Just pass the flame along the plate and bubbles should surface and pop giving a bubble-free plate.
0 notes
Text
LB Agar plates
Few months ago I determined that my strain of interest fluoresces best in M9 media which like DMEM is a minimal media unlike LB. So I had been using glycerol stocks to inoculate M9 media for 4hr cultures. This never gave enough bacteria to do reproducible studies on. One of my lab-mates suggested that the cleanest way to grow bacteria even of M9 is the media for experiments is to:
1. Streak glycerol stock on LB agar plate with Kanamycin (or your relevant antibiotic for selectivity). After overnight growth, the plates are good for 1-2 weeks at 4C.
2. Take colonies on the agar plate to inoculate M9 media. This give the bacteria enough time in rich media to reach a productive stage before being put in minimal media.
I streaked the LB agar plate this afternoon and hope to see good colonies tomorrow.
0 notes
Text
FRAP
Today I learned a neat technique called Fluorescence Recovery After Photobleaching which does exactly what the name says. You select a region of the sample and focus a laser beam on it. This bleaches the region. The recovery of fluorescence in the region post-bleaching tells you
1. The cells in the region are still alive
2. Time to near-complete recovery gives information on diffusion rates (specifically in the case of lipid bilayers).
0 notes
Text
Epigenetics
Genetic variations in the same specie where the outcome of different genetic pathways is the same but the route taken is different. From Wikipedia;
http://en.wikipedia.org/wiki/Epigenetics
Robin Holliday defined epigenetics as "the study of the mechanisms of temporal and spatial control of gene activity during the development of complex organisms."[8] Thus epigenetic can be used to describe anything other than DNA sequence that influences the development of an organism.
0 notes
Text
Leica DMIRE 2
I recently found out Leica DMIRE 2 can take laser excited fluorescence and brightfield images at the same time. This is fantastic since I can now take pictures like these;
0 notes
Text
Preparing a 10% Succinic acid solution
Yesterday while making the M9 growth media, I had a bit of a trouble dissolving 4gms of Succinate in 40ml of water since its solubility limit at room temperature is 58gm/L which corresponds to about 2.3gm/40mL. To overcome the problem my lab mate suggested that I drop a few pellets of KOH which we didn't have. So I found NaOH instead which worked perfectly well.
0 notes