#IEC 62304
Text
Things You Need to Know About Medical Device Software Validation
Software Verification and Validation (Software V&V) is an integral part of software design that spans all the development stages as specified in IEC 62304 which addresses the Software Development Life Cycle (SDLC) of medical software and software embedded within medical devices. Software Verification is to test if the software was designed as per requirements and Software Validation is to test if the right product was built for the user. Medical device software validation generally occurs during or at the end of the development cycle.
The difference between Software Validation & Software Verification can be answered by asking these mentioned questions:
· Verification: Are we building the product, right?
· Validation: Are we building the right product?
Software validation is a process of checking if the product will meet the customer’s actual needs, while verification involves procedures for making certain that the software is well-engineered, free of errors, and functional.
Following are the steps for Software Validation for Medical Devices
· Create a software validation plan
· Determine system requirements
· Create a validation protocol and test specifications
· Conduct and document tests
· Establish procedures and write your final report
IZiel has highly trained software engineers with multiple years of experience in software coding, software verification and software validation. The team consists of senior engineers who have worked in the design and development of highly sophisticated implantable devices at industry-leading companies, with direct expertise in software V&V.
0 notes
Text
Tag game
tagged by @the-cooler-sidestep thank youuu uwu
Last song; Granite by Sleep Token (I am so normal about this band. SO. NORMAL.)
Currently watching; binging a bunch of NeoCranium vods because A: gay for robots/nonhuman B: is veri funni
Currently reading; ISO 27001 / 13485 and IEC 81001-5-1 / 62304. because I only have energy to read for work bullshit these days :))
Current obsession; troubleshooting my jank ass gaymer PC that crashes/freezes/bluescreens constantly while I just wanna play the closed beta for The Finals before it's over :')
all of my mutuals are fucking hottie cuties so you're all tagged if you want to be uwu, but I'mma also just tag the ones that come to mind
@kermakatti @korppuhiiri @haurgrimr @doegirldick @diamantefangs @zondearts
3 notes
·
View notes
Text
IMDRF Regulation on SaMD | OMC Medical Limited

The SaMD definition statement should include a clear and robust statement about intended use, including the following:
https://jpcdn.it/img/r/929/656/909ac9463cfdf9c186eef253a05c015d.png
SaMD is software that carries out one or more medical tasks. If it might be incorporated with a piece of hardware, the software performs a medical function.
The international medical device regulators Forum (IMDRF) defines SaMD as “software that may operate on general-purpose (non-medical) computing platforms, may be combined with other products, including medical devices, and may interface with other medical devices or other general-purpose hardware or software that provide input to SaMD.”
Software that turns the magnets in an MRI or animates an X-ray control panel are examples of software that is necessary for hardware operation but is exempt from SaMD. Software that only organizes data, retrieves data, or speeds up operations is also insufficient.
7 Classification of IMDRF Regulation on Software as a Medical Device
The following are necessary principles required in the categorization approach of SaMDs.
An accurate SaMD definition is a prerequisite for the categorization of the device.
The importance of the information provided by the SaMD to the healthcare decision and the situation or condition are combined to determine the categories.
The four classifications (I, II, III, and IV) are based on the severity of the impact on the patient or public health, where accurate information provided by the SaMD to treat or diagnose, guide or inform clinical management is essential to prevent death, long-term disability, or other severe deterioration of health, mitigating public health.
The impact level is at its maximum in Category IV and lowest in Category I.
According to the details provided in the SaMD definition statement, a manufacturer’s SaMD is classified in the highest category when it can be employed in various healthcare settings or conditions.
The classification of SaMD should be correctly re-evaluated when a manufacturer modifies SaMD during the lifecycle that causes the definition statement to change. The information in the revised (new) SaMD definition statement is used to categorize the SaMD.
Even when a SaMD is interfaced with other SaMDs, other hardware medical devices, or employed as a module in a more complex system, it falls under the category specified in its SaMD defining statement.
State of Healthcare situation or conditionSignificance of information provided by SaMD to healthcare decision Treat or diagnoseDrive clinical managementInform clinical managementCriticalIVIIIIISeriousIIIIIINon-seriousIIII
The above table represents the risk categorization of IMDRF regulation for SaMD based on the significance of the information provided by SaMD to healthcare decisions and the state of the healthcare decision.
Identifying risks and putting safeguards in place that assure acceptable risks are necessary for creating safe SaMD. It is well acknowledged that testing software is insufficient to establish operational safety.
As a result, it is acknowledged that confidence should be included in the software to guarantee its security.
The IEC 62304 standard governs the lifecycle development of medical device software. The standard highlights three key safety concepts that are pertinent to SaMD, defines basic testing requirements, and proposes a risk-based decision methodology:
Risk management.
Quality management.
Methodical and systematic systems engineering according to best industry practices.
Combining these concepts helps IMDRF regulation on SaMD manufacturers follow a clearly structured and consistently repeatable decision-making process to promote safety for SaMD.
Design and Development
A quality-assured approach to software development should consider the selection and implementation of system design and development methods that:
Incorporate a structured development using models, methods, architecture and techniques appropriate for the development language(s) and the device’s intended use,
Cover the various stages of the software lifecycle using IEC 62304 as a reference standard for software development. Software Engineering Body of Knowledge (SWEBOK) and Systems Engineering Body of Knowledge (SEBoK) must be used as reference books for software engineering and
The design and development process must be documented systematically (using appropriate tools.)
Post Market Surveillance
SaMD manufacturers should continuously monitor customer complaints to maintain the safety level because software hazards can never be eradicated.
Customer feedback must be gathered as part of the monitoring process, for instance, through enquiries, grievances, market research, focus groups, service, etc. SaMD and other software have inherent characteristics that effectively comprehend and record user experiences.
The use these feedback mechanisms by IMDRF regulation on SaMD makers is advised to comprehend failure modes and conduct analysis to address safety concerns.
Additionally, it is recommended that SaMD manufacturers expand their monitoring to detect system or software errors automatically, i.e., identify and fix an error before a failure happens.
Quality Management System
Medical device QMS principles allow the measurement of activities depending on
The type of medical device.
Risk of the product to patients.
Size of the organization.
Technology or automation is used to manufacture.
And other factors are determined by the manufacturer to control quality and maintain the safe and effective performance of the medical device.
https://jpcdn.it/img/r/799/509/da3a73838ee9d6f6bc1d38e8c9ed5eaa.png
SaMD, a software-only product, is manufactured primarily using automated software development tools to assist development lifecycle activities.
In some circumstances, these computerized processes could take the role of deliberate or discrete acts generally present in creating physical items (such as transferring design to production).
To control the quality of SaMD, however, it is still necessary to adhere to the QMS principles that give the lifecycle processes and activities structure and support.
An effective QMS for IMDRF regulation on SaMD have to include the following principles:
An organizational structure ensures SaMD’s safety, efficacy, and performance by providing leadership, accountability, and governance with enough resources.
A set of SaMD lifecycle support processes that are scalable to the organization’s size and applied consistently across all realizations and use processes
A set of realizations and use processes that are scalable for the type of SaMD and the organization’s size takes into account essential elements required for assuring the safety, effectiveness, and performance of IMDRF regulation on SaMD.
Originally Published at: https://omcmedical.com/imdrf-regulation-on-samd/
0 notes
Text

Healthcare App Development Company
Wi4 has years of experience working with mobile medical app developers to ensure your product is secure for providers and patients. We will work closely together so you can get started on the right foot by meeting all applicable regulatory requirements from day one!
We will take care of the following compliance requirements:
-> GDPR Compliance
-> HIPAA Compliance
-> IEC 62304 Compliance for Medical Device Apps
Email us: [email protected]
Call: (470) 830-0294
Visit: 855 Peachtree St NE, Unit 1707, Atlanta, GA 30308, USA
Website: https://wi4.org/services/healthcare-mobile-app-development-company/
HealthTech #healthcareapp #MedicalApps #Innovation #DigitalHealth #wi4corporation #vineetagrawal #atlanta #georgia
0 notes
Text
De Novo paving the path for Technological Advancements

While there existed many computerized and digital tomographs with specific applications (Trauma CT, Cardiac CT, CT used in Nuclear Medicine / PET), there has never been one with an application in the respiratory space. Electrical impedance tomography (EIT) is a noninvasive, non-radiologic imaging modality that is useful for the assessment of lung disorders during mechanical ventilation. EIT offers potentially important benefits over standard imaging modalities since it is portable in nature and has non-radiological characteristics, which makes it conducive for medical use and diagnostic applications.
Device Classification
🔘 According to the FDA, any new device which is not in commercial distribution will be automatically classified as Class III devices irrespective of the level of risk associated with the device. This comes after the post amendment of FD&C Act (Medical Devices Act), May 28, 1976.
🔘 Since Electrical Impedance Tomograph (EIT) is a non- commercial device developed after May 28, 1976, it is classified as a Class III device. Since no predicates existed with regard to ventilator electrical impedance tomograph, the next step was to go for a De Novo Classification.

Devices can be classified into Class I or Class II under section 513(i) of the FD&C Act i.e. the reclassification of devices.
Devices Reclassification by De Novo Process
The De Novo process provides a pathway to classify novel medical devices for which general controls alone, or general and special controls, provide reasonable assurance of safety and effectiveness for the intended use, but for which there is no legally marketed predicate device. De Novo classification is a risk-based classification process.
De Novo Classification
De Novo classification is beneficial to the consumers as they can gain access to innovative devices which go through just the right amount of regulatory rigour.
Timpel Inc. is the company which is registered under De Novo for reclassification of Ventilator Electrical Impedance Tomograph device. It is the first of its kind that cleared De Novo and is now approved by the FDA as a Class II device under the Device Classification Name: Ventilator Electrical Impedance Tomograph.
The features of this device are listed as follows:
➡️ Intuitive: Integrated clinical decision support tools
➡️ Realtime: High temporal resolution
➡️ Easy to install, simple to use
➡️ Portable, can benefit multiple patients
➡️ Comfortable ergonomic belts
➡️ Non-Invasive in nature
➡️ Portable for bed-side access, no relocation of patient is required
➡️ Radiation Free: Can be used frequently
➡️ Provides continuous, real-time images
➡️ Operator independent (supervision not necessary)
Special Control Tests for De Novo Classification for Class II devices
Since the device includes a wide variety of features, the special controls of the FDA were considered for the device to ensure that the device is safe and effective for human use. Technical tests according to amended 21 CFR part 868 (Anesthesiological Devices) are mentioned in the table below along with the standards.
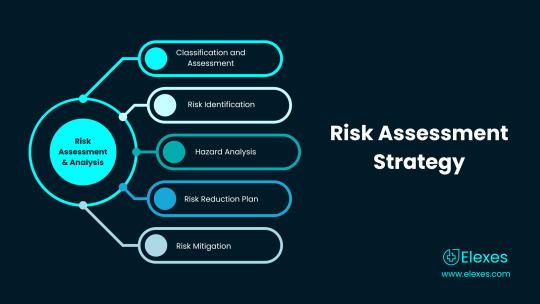
Risk and Mitigation steps associated with the device
Tissue Reaction (Adverse in nature) - Biocompatibility testing/evaluation (ISO 10993-1:2018)
Electromagnetic Interferences - Electromagnetic Compatibility Testing (ISO 60601-1: 2018)
Communicable/Non Communicable infections - Proper Packaging and labeling implementation (ISO 16142-1:2016)
Image distortion due to hardware and software malfunction - Software verification and Validation (IEC 62304:2006)Risk Analysis (ISO 14971:2007)Non Clinical Performance Testing (60068-2:2018
Thermal and/or electrical shock injury - Mechanical and Thermal safety Testing (ISO 60068-2)
Additional tests to be carried out under De Novo are:
Guidance for image interpretation
Instructions for reprocessing
Plethysmography accuracy testing
Benefits of De Novo Classification
Upon getting approved by the FDA under De Novo classification, the device was assigned as the generic type ‘Ventilator Electrical Impedance Tomograph’ and was defined as a non-radiological and non-invasive ventilator device that provides an assessment to the variations in local impedances within a cross-section of a patient’s thorax. This is quite a remarkable achievement due to the following reasons:
This De Novo approval of Timple Inc. paves the path for bringing more such innovative Ventilator Tomographs to the market.
Similar devices in the future shall have a lesser regulatory burden and can be easily accessed by the users.
The device can now be used as a predicate by other manufacturers, making similar device, for 510(k) clearance of their products which means the manufacturers do not have to submit a separate De Novo or Pre Market approval form for their products.
The De Novo classification has reclassified the device as a Class II device with special controls thus making it self-sufficient in mitigating the residual risks, since the general control functions cannot mitigate some of the specific residual risks.
Conclusion
De Novo classification is not just a device reclassification process for medical devices but it is an application that the manufacturers can use in order to commercialize their products.
This process clearly enables the manufacturer to know about their products genuinity and reduces the regulatory burdens which are commonly associated with other classification processes.
The effort of one can lessen the efforts of different manufacturers for their devices. Also, these manufacturer’s can pursue new angles of modifications and improvement for better accessibility and treatment opportunities.
To do a DeNovo certification the sponsor can wait for the FDA to get back stating not substantially equivalent (NSE) determination in response to a 510(k) submission or the manufacturer can determine that there is no legally marketed predicate device yet, to produce substantial equivalence and apply for a DeNovo.
Sponsor’s or Manufacturers can also contact professional service firms like Elexes Medical Consulting to acquire the assistance for such submissions and obtaining an approval from the FDA. Feel free contact [email protected] with any questions or comments on this content.
1 note
·
View note
Text
IAR fully supports Infineon's latest TRAVEO T2G CYT6BJ body control MCU family products

According to the information received by Lansheng Technology, it has fully supported the latest CYT6BJ series in Infineon's TRAVEO™ T2G body control MCU family. IAR Embedded Workbench for Arm is a complete embedded development solution, equipped with highly optimized compiler and build tools, code analysis tools C-STAT and C-RUN, and powerful debugging functions. This enables developers working on complex automotive body electronics applications to take full advantage of the features of the TRAVEO™ T2G MCU to create innovative designs with high code quality. IAR Embedded Workbench for Arm supports AUTOSAR and provides a functional safety version to help customers accelerate product certification.
Clara Volkmar, Director Product Marketing Body and Driver Information, Infineon's Automotive Division, said: "Infineon's TRAVEO™ T2G microcontrollers provide a compact solution to meet the needs of modern automotive body electronics systems and can take advantage of IAR Professional and suitable third-party development tools such as Embedded Workbench for Arm to achieve efficient development. Thanks to such a powerful tool partner as IAR, developers can quickly realize their demanding automotive applications even with our latest CYT6BJ series chips project."
By supporting all available TRAVEO™ T2G series chips, including the latest CYT6BJ series, IAR Embedded Workbench for Arm ensures compatibility and provides developers with highly optimized build tools and advanced debugging features: such as complex code and data interrupts Points, runtime stack analysis, call stack visualization, code coverage analysis, and integrated monitoring of power consumption contribute to a seamless development experience. With the code analysis tools C-STAT and C-RUN, developers can fully control the code quality.
For companies with functional safety requirements, IAR Embedded Workbench for Arm offers a functional safety version certified by TÜV SÜD (certified according to ISO 26262 requirements). This functional safety version is also certified according to IEC 61508, IEC 62304, EN 50128, EN 50657, IEC 60730, ISO 13849, IEC 62061, IEC 61511 and ISO 25119. For companies using continuous integration (CI) workflows and automated build and test processes, IAR Build Tools is also available for Linux-based frameworks. In addition, IAR's professional technical support, training and flexible licenses enable all customers to find a solution that suits their specific needs.
Lansheng Technology Limited, which is a spot stock distributor of many well-known brands, we have price advantage of the first-hand spot channel, and have technical supports.
Our main brands: STMicroelectronics, Toshiba, Microchip, Vishay, Marvell, ON Semiconductor, AOS, DIODES, Murata, Samsung, Hyundai/Hynix, Xilinx, Micron, Infinone, Texas Instruments, ADI, Maxim Integrated, NXP, etc
To learn more about our products, services, and capabilities, please visit our website at http://www.lanshengic.com
0 notes
Text
Embedded Software Manufactory (31/08/2021)
RTOS claims to be IEC 62304 compliant receives warning from medical device regulators
... In this article, we have covered blackberry QNX, which is shipped by Japanese distributor Fujisoft with a certificate of compliance with IEC 62304 (Medical Equipment Software-Software Lifecycle Process) Software Safety Class C. I wanted to show the mistake that this deviated from the idea of the risk-based approach of IEC 62304 and was merely using the standard to sell merchandise.
1 note
·
View note
Photo

#IEC #62304 #ISO #13845 #Functional #Safety #Medical #Devices #Medical #Software #Development #Lifecycle #Quality #Management https://www.instagram.com/p/CbvW2MfK63f/?utm_medium=tumblr
#iec#62304#iso#13845#functional#safety#medical#devices#software#development#lifecycle#quality#management
0 notes
Text
Demystifying Medical Device Software Validation: Ensuring Safety and Compliance
Briefly introduce the importance of medical device software validation.
Explain that the blog post will provide an overview of the key concepts, processes, and regulations related to medical device software validation.
I. Understanding Medical Device Software
Define what medical device software is and its significance in healthcare.
Discuss the different types of medical device software (e.g., embedded, standalone, mobile apps).
Emphasize the increasing reliance on software in modern medical devices.
II. Why Software Validation Matters
Explain the critical role of software validation in ensuring patient safety.
Discuss the potential risks and consequences of inadequate software validation.
Highlight real-world examples of medical device software failures and their implications.
III. Regulatory Framework
Introduce the regulatory bodies and standards governing medical device software validation (e.g., FDA, ISO 13485, IEC 62304).
Explain the classification of medical devices and how it affects validation requirements.
Provide an overview of the FDA's Software as a Medical Device (SaMD) guidance.
IV. The Software Validation Process
Outline the key steps in the software validation process:
Requirements analysis and specification
Risk assessment and management
Design and development
Testing and verification
Validation and documentation
Emphasize the importance of traceability and documentation.
V. Common Challenges and Solutions
Discuss common challenges in medical device software validation (e.g., changing requirements, evolving technology).
Provide practical solutions and best practices for overcoming these challenges.
VI. Case Studies
Present real-world case studies of successful medical device software validation projects.
Highlight the benefits of proper validation, including improved patient outcomes and reduced liability.
VII. Future Trends in Medical Device Software Validation
Explore emerging trends and technologies in medical device software validation (e.g., artificial intelligence, machine learning).
IZiel has highly trained software engineers with multiple years of experience in software coding, software verification and software validation. The team consists of senior engineers who have worked in design and development of highly sophisticated implantable devices at industry leading companies, with direct expertise in software V&V. This team, with the support of IZiel’s regulatory and clinical experts, are decidedly equipped to handle complex software validation activities for medical device manufacturers. Integrating risk assessments into the validation lifecycle and documenting the basis for what was done also provides a level of assurance to management and regulatory authorities that the system was properly defined, designed, built, tested, operated, and maintained.
0 notes
Text
Clara Holoscan MGX de Nvidia significa llevar IA de alta potencia al consultorio médico
Nueva Noticia publicada en https://noticiasq.com/clara-holoscan-mgx-de-nvidia-significa-llevar-ia-de-alta-potencia-al-consultorio-medico/
Clara Holoscan MGX de Nvidia significa llevar IA de alta potencia al consultorio médico
Esta semana, Nvidia, una empresa mejor conocida por sus unidades de procesamiento gráfico (GPU) de alta potencia, presentó una plataforma para el desarrollo de dispositivos médicos impulsados por IA. El dispositivo, llamado Clara Holoscan MGX, proporciona potencia informática que permite a los sensores médicos procesar múltiples flujos de datos en paralelo, entrenar algoritmos de IA y visualizar la biología en tiempo real.
Clara Holoscan MGX, debutó en la conferencia GTC 2022 de Nvidia, es una «plataforma de robótica abierta y escalable», como lo expresó el CEO Jensen Huang en un discurso de apertura. Es una pila de hardware y software diseñada para ayudar a conectar sensores o dispositivos médicos robóticos con aplicaciones de IA.
¿Cómo podría funcionar eso? Tome el proceso de endoscopia como un ejemplo. Por lo general, un médico insertará una pequeña cámara dentro de su cuerpo y observará a su alrededor. Clara Holoscan MGX puede conectarse directamente a esa cámara y procesar en tiempo real los datos que se recopilan. Luego, esos datos podrían introducirse en modelos de IA que podrían detectar anomalías, navegar a través de su anatomía y ayudar al cirujano a elaborar un plan de tratamiento. (Para ser claros, estos modelos de IA no serían fabricados por Nvidia, solo se ejecutarían en su hardware).
Nvidia ya es conocida por sus GPU, que son especialmente buenas para ejecutar rápidamente cálculos en paralelo. Las GPU alguna vez fueron más conocidas por los jugadores, pero se han convertido en un acelerador clave para cualquier industria interesada en entrenar redes neuronales profundas. Las redes neuronales profundas necesitan analizar rápidamente miles de millones de puntos de datos a medida que aprenden, por ejemplo, a leer una radiografía. Y los modelos resultantes también necesitan una gran cantidad de computación para usarse en tiempo real, lo cual esta plataforma pretende proporcionar.
youtube
Nvidia es ahora un jugador dominante en el espacio de la IA porque proporciona la fuerza computacional bruta necesaria para muchos de estos proyectos, y lo ha hecho fácil con una flota de combinaciones de hardware y software específicas de la industria. Por ejemplo, Nvidia ha estado activa en el espacio de los automóviles autónomos con proyectos como Nvidia Drive, una plataforma para la formación y construcción de vehículos autónomos.
Nvidia ya ha comenzado a hacer propuestas en el espacio de la atención médica. La plataforma Clara se anunció por primera vez en 2018 y se diseñó inicialmente para crear una experiencia de imágenes médicas fluida. La plataforma se ha ampliado a lo largo de los años, pero la plataforma Clara Holoscan MGX está destinada básicamente a convertirse en una ventanilla única.
Kimberly Powell, vicepresidenta de atención médica de Nvidia, dijo a TechCrunch que Clara Holoscan es «una plataforma completa de extremo a extremo». Nvidia Clara Holoscan es para los dispositivos médicos lo que Nvidia Drive es para los vehículos autónomos”, dijo.
Powell dice que las principales innovaciones de Clara Holoscan son dos. Primero, ha sido diseñado para cumplir con el estándar IEC 62304, que es un proceso de referencia para el desarrollo seguro de software médico. Entonces, básicamente está repleto de lo que Huang llama una cantidad «increíble» de poder de cómputo.
Juntas, la combinación debería permitir que las empresas que buscan construir o entrenar dispositivos médicos alimentados por IA avancen mucho más rápido.
Créditos de imagen: nvidia
“La arquitectura de Clara Holoscan reduce significativamente la inversión en ingeniería necesaria para llevar al mercado un nuevo dispositivo médico o software como dispositivo médico”, dijo Powell.
Ya hay muchas empresas que buscan hacer exactamente lo que Nvidia propone: combinar dispositivos e IA. Activ Surgical, por ejemplo, una startup que trabaja en endoscopios quirúrgicos asistidos por IA (llamados ActivSight) ya usa la GPU de Nvidia y está trabajando en más aplicaciones de IA informadas por datos de endoscopio. Para ello, la empresa fue aceptada en el programa Inception de Nvidia, que le dio acceso anticipado al kit de desarrollo Clara AGX. Ese kit, según un comunicado de prensa, se hizo eco de la afirmación de Powell de que la tecnología de Nvidia puede acelerar el desarrollo de productos.
“El kit de desarrollo también reducirá el tiempo de desarrollo general para sacar al mercado los futuros productos de Activ Surgical, incluido ActivSight, en los próximos dos años”, se lee en el comunicado de Activ Surgical.
Por el momento, los poderes completos de Clara Holoscan no están disponibles. Durante su discurso de apertura, Huang dijo que la tecnología de grado médico no estará disponible para acceso anticipado hasta el primer trimestre de 2023. En ese momento, agregó Powell, los socios ODM de Nvidia establecerán los precios del hardware y los precios del software «estarán disponibles». (Es probable que esa información aparezca aquí).
Por ahora, el lanzamiento de Clara Holoscan MGX parece un refuerzo del punto de apoyo ya firme de Nvidia en el espacio de atención médica de IA. Básicamente, está construyendo la base computacional que se encuentra debajo.
Y ese es un buen lugar para estar. Según el informe del Índice de IA de 2022 de Stanford, las dos mayores áreas de inversión privada en IA se encontraban justo en este punto de mira en 2021: $ 12,2 mil millones en gestión de datos, procesamiento y computación en la nube, y $ 11,29 mil millones en herramientas médicas y de atención médica.
0 notes
Text

Unlock the Key to App Compliance with Wi4! 🌐
At Wi4, we've got your back when it comes to ensuring your medical app meets all the compliance requirements. Whether it's HIPAA, GDPR, or IEC 62304, we've got you covered!
HIPAA Compliance ✔️
GDPR Compliance ✔️
IEC 62304 Compliance ✔️
Trust us to make sure your app is secure for both providers and patients while meeting all necessary regulatory standards. Don't risk hefty fines and penalties – stay compliant with Wi4.
Learn More: https://wi4.org/services/healthcare-mobile-app-development-company/
Call (470) 830-0294
HealthcareApps #Compliance #MedicalApp #HIPAA #GDPR #IEC62304 #AppDevelopment #Wi4
1 note
·
View note
Text
Why is compliance with regulatory requirements important for Medical Devices testing?

The role of medical devices in screening, diagnostics, and treatment has become critical. Modern medical devices have sophisticated components with digital interfaces. These help to derive meaningful inferences from the data emanating from patients’ vital stats. Since speed and quality lie at the core of the functioning of medical devices, they need to comply with quality standards and regulations. Also, since the functioning of medical devices is regulated by the built-in software, the same should adhere to basic safety guidelines or protocols.
Further, the rate of failure of medical devices has been found to be increasing by the day leading to the establishment of IEC 62304.
What is IEC 62304?
It is the international regulatory standard defining the SDLC requirements of software driving medical devices. The standard was established as it was felt that product testing alone will not ensure the safety of patients, especially when there is a software present. The standard requires every aspect of the SDLC to be scrutinized. These include development, configuration, risk management, maintenance, security, and problem resolution.
IEC 62304 offers a standard framework for the manufacturers of medical devices to design software. By conforming to this standard, manufacturers can fulfil the requirements of medical devices testing thereby generating trust. Also, since the standard is harmonized with the medical device directive practiced in the European Union, it has been acknowledged as a benchmark. The devices adhering to IEC 62304 include
Diagnosing, monitoring, or treating patients under medical supervision
Contacting the patient - physical or electrical
Transferring energy to/from the patient
Monitoring or detecting such energy transfer
Digitization in healthcare can have many dimensions. These include monitoring the performance of applications, leveraging the digital ecosystems for the stakeholders, and calculating the quantum of investments to drive digital transformation. The healthcare sector is being transformed with the infusion of new technologies (wearables included) and treatment/diagnostics methodologies. Since devices incorporating new technologies need to deliver results with precision, they should undergo rigorous medical devices testing. Let us discuss the reasons for validating medical devices through comprehensive Quality Assurance.
Security: Medical devices contain sensitive data about patients’ health, which if breached, can lead to severe consequences. It is only through rigorous healthcare software testing that devices can be made hack-proof. The measures may include identifying vulnerabilities, validating and authenticating user log-ins, performing penetrating testing against firewalls, or encrypting data. Also, medical devices need to adhere to stringent quality standards such as the Health Insurance Portability and Accountability Act (HIPAA). This ensures the protection of patients’ health-related data and information.
Usability: This is a crucial testing requirement as devices are handled by healthcare professionals while discharging their duties. Since many professionals may find it difficult to handle the features and functionalities of devices, the same should be made simpler. This is where usability testing using automation can help to simplify and enhance the user experience.
Big data: The healthcare sector deals with a humongous quantum of data based on which inferences are drawn about the health condition of patients. These inferences are further leveraged to plan the right treatment strategy or develop a product. Big data analytics can help to derive the right inferences from the data quickly and accurately. It can help professionals to make informed decisions related to research and development, drug inventions, or curing ailments.
Device interoperability: Medical devices need to connect and interoperate to deliver the required outcome and user experience. Since the healthcare sector needs to ensure data privacy, security, and regulatory compliance, the role of medical devices testing specialists becomes crucial. So, healthcare testing services need to apply technical expertise, resources, and time to ensure quality, compliance, and business profitability.
The testing of medical devices impinges on meeting the regulatory requirement. Moreover, navigating the regulatory ecosystem is crucial for the successful launch of medical devices or products. It ensures the adherence of devices to attributes like performance, safety, and security. Also, with the increased complexity of treatment protocols, the standards of quality and safety of medical devices should be enhanced. These include adherence to Electromagnetic Compatibility (EMC) testing for devices with power supply and electronic components. The quicker these standards are complied with, the faster companies can expect their products to become market-ready.
Conclusion
The healthcare sector deals with patients’ data and information on a daily basis. To ensure the security of these, the medical devices testing experts ought to ensure interoperability and flawless performance of such devices. This is where adherence to regulatory protocols becomes important to deliver better customer experience and market adoption.
This article is already published on dev.to.
#Medical devices testing#Healthcare software testing#healthcare testing services#medical devices testing specialists#medical devices testing experts
0 notes
Text
Elektronik Praxis (13/07/2021)
Wann ist eine Software ein Medizinprodukt?
... Anforderungen an die Bedienbarkeit definiert die internationale Norm IEC 62366-1. Oft wird das Risiko unterschätzt, das von einer mangelnden Benutzerfreundlichkeit ausgeht.
... Hersteller sollten ebenfalls die international gültige Norm IEC 62304 berücksichtigen. Sie stellt Mindestanforderungen an die wichtigsten Prozesse im Lebenszyklus einer Software. Das reicht von der Entwicklung über die Wartung bis hin zum Risikomanagement und zur Problemlösung.
0 notes
Photo

Medical Device Software Development
Camensys medical device software development compliant to ISO 13485 and IEC 62304 standard. Camensys offers services for devices that are categorized by the FDA or SaMD.
0 notes
Text
Latest Release of VxWorks Cert Edition IEC 62304 Compliant for Medical Device Development
Latest Release of VxWorks Cert Edition IEC 62304 Compliant for Medical Device Development

ALAMEDA, Calif.–(BUSINESS WIRE)–#IEC62304—Wind River®, a leader in delivering software for the intelligent edge, today announced IEC 62304 medical standard compliance for the latest release of VxWorks® Cert Edition. The real-time operating system (RTOS) for safety-critical applications is designed and developed to the highest achievable safety levels accepted by worldwide certification…
View On WordPress
0 notes
Text
De Novo paving the path for Technological Advancements
While there existed many computerized and digital tomographs with specific applications (Trauma CT, Cardiac CT, CT used in Nuclear Medicine / PET), there has never been one with an application in the respiratory space.
Electrical impedance tomography (EIT) is a noninvasive, non-radiologic imaging modality that is useful for the assessment of lung disorders during mechanical ventilation.
EIT offers potentially important benefits over standard imaging modalities since it is portable in nature and has non-radiological characteristics, which makes it conducive for medical use and diagnostic applications.
Device Classification
According to the FDA, any new device which is not in commercial distribution will be automatically classified as Class III devices irrespective of the level of risk associated with the device. This comes after the post amendment of FD&C Act (Medical Devices Act), May 28, 1976.
⊙ Since Electrical Impedance Tomograph (EIT) is a non- commercial device developed after May 28, 1976, it is classified as a Class III device. Since no predicates existed with regard to ventilator electrical impedance tomograph, the next step was to go for a De Novo Classification.
Devices can be classified into Class I or Class II under section 513(i) of the FD&C Act i.e. the reclassification of devices.
Devices can be reclassified by:
De Novo Process
The De Novo process provides a pathway to classify novel medical devices for which general controls alone, or general and special controls, provide reasonable assurance of safety and effectiveness for the intended use, but for which there is no legally marketed predicate device. De Novo classification is a risk-based classification process.
De Novo Classification
De Novo classification is beneficial to the consumers as they can gain access to innovative devices which go through just the right amount of regulatory rigour.
Timpel Inc. is the company which is registered under De Novo for reclassification of Ventilator Electrical Impedance Tomograph device.
It is the first of its kind that cleared De Novo and is now approved by the FDA as a Class II device under the Device Classification Name: Ventilator Electrical Impedance Tomograph
The features of this device are listed as follows:
➩ Intuitive: Integrated clinical decision support tools
➩ Realtime: High temporal resolution
➩ Easy to install, simple to use
➩ Portable, can benefit multiple patients
➩ Comfortable ergonomic belts
➩ Non-Invasive in nature
➩ Portable for bed-side access, no relocation of patient is required
➩ Radiation Free: Can be used frequently
➩ Provides continuous, real-time images
➩ Operator independent (supervision not necessary)
Special Control Tests for De Novo Classification for Class II devices
Since the device includes a wide variety of features, the special controls of the FDA were considered for the device to ensure that the device is safe and effective for human use.
Technical tests according to amended 21 CFR part 868 (Anesthesiological Devices) are mentioned in the table below along with the standards.
Risk and Mitigation steps associated with the device
➩ Tissue Reaction (Adverse in nature) – Biocompatibility testing/evaluation (ISO 10993-1:2018)
➩ Electromagnetic Interferences – Electromagnetic Compatibility Testing (ISO 60601-1: 2018)
➩ Communicable/Non Communicable infections – Proper Packaging and labeling implementation (ISO 16142-1:2016)
➩ Image distortion due to hardware and software malfunction – Software verification and Validation (IEC 62304:2006)Risk Analysis (ISO 14971:2007)Non Clinical Performance Testing (60068-2:2018
➩ Thermal and/or electrical shock injury – Mechanical and Thermal safety Testing (ISO 60068-2)
Additional tests to be carried out under De Novo are:
Guidance for image interpretation
Instructions for reprocessing
Plethysmography accuracy testing
Benefits of De Novo Classification
Upon getting approved by the FDA under De Novo classification, the device was assigned as the generic type ‘Ventilator Electrical Impedance Tomograph’ and was defined as a non-radiological and non-invasive ventilator device that provides an assessment to the variations in local impedances within a cross-section of a patient’s thorax.
This is quite a remarkable achievement due to the following reasons:
⊙ This De Novo approval of Timple Inc. paves the path for bringing more such innovative Ventilator Tomographs to the market.
⊙ Similar devices in the future shall have a lesser regulatory burden and can be easily accessed by the users.
⊙ The device can now be used as a predicate by other manufacturers, making similar device, for 510(k) clearance of their products which means the manufacturers do not have to submit a separate De Novo or Pre Market approval form for their products.
⊙ The De Novo classification has reclassified the device as a Class II device with special controls thus making it self-sufficient in mitigating the residual risks, since the general control functions cannot mitigate some of the specific residual risks.
Bottom Line
De Novo classification is not just a device reclassification process for medical devices but it is an application that the manufacturers can use in order to commercialize their products.
This process clearly enables the manufacturer to know about their products genuinity and reduces the regulatory burdens which are commonly associated with other classification processes.
The effort of one can lessen the efforts of different manufacturers for their devices. Also, these manufacturer’s can pursue new angles of modifications and improvement for better accessibility and treatment opportunities.
To do a DeNovo certification the sponsor can wait for the FDA to get back stating not substantially equivalent (NSE) determination in response to a 510(k) submission or the manufacturer can determine that there is no legally marketed predicate device yet, to produce substantial equivalence and apply for a DeNovo.
Sponsor’s or Manufacturers can also contact our professionals to acquire the assistance for such submissions and obtaining an approval from the FDA. Feel free contact at [email protected] for any questions or comments on this content.
0 notes
Last Seen Blogs

steveroqers-m
CAPTAIN

panindiacarrental
Untitled

massillonlibrary208
Massillon Public Library

ma-athan
draw and foto
