#multistep mutation
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
The Tumblr app for Google Glass was released on May 16, 2013.
Text
Neo-Darwinism Must Mutate to Survive
No. 5 Story of 2023: Peer-Reviewed Paper Finds “Neo-Darwinism Must Mutate to Survive”
View On WordPress
0 notes
Text
Dr. von Ardenne on Cancer, Inflammation and Oxygen
Developed in the late 1960s by Professor von Ardenne, (a student of Dr. Otto Warburg, best known for his pioneering research on the connection between lack of oxygen and cancer), Oxygen Multistep Therapy combines oxygen therapy, drugs that facilitate intracellular oxygen turnover, and physical exercise adapted to individual performance levels. This unique therapy has diversified into more than 20 different treatment variants and is now practiced in several hundred settings throughout Europe. Ardenne put his finger on how inflammation interferes with oxygen transfer to cells.
“It is believed that cancer is caused by an accumulation of mutations in cells of the body,” says Dr. Carlo M. Croce, professor and chair of molecular virology, immunology and medical genetics. “Our study suggests that miR-155, which is associated with inflammation, increases the mutation rate and might be a key player in inflammation-induced cancers generally.” This and many other studies show how inflammation can help cause cancer. Chronic inflammation due to infection or to conditions such as chronic inflammatory bowel disease is associated with up to 25% of all cancers. Manfred von Ardenne (20 January 1907 – 26 May 1997) was a German researcher, applied physicist and inventor. He holds approximately 600 patents in fields including electron microscopy, medical technology, nuclear technology, plasma physics, and radio and television technology.
Oxygen Multi-Step Therapy has become more commonly known as Exercise with Oxygen Therapy (EWOT). Although there are different ways to practice EWOT, the core of Dr. von Ardenne’s therapeutic practice is the breathing of pure oxygen while exercising. This allows additional oxygen to be absorbed by your red blood cell, blood plasma and tissue fluids. Professor Ardenne wrote[1], “Because more than 80% of all cancer deaths are caused by metastases, development and evaluation of methods for fighting tumor dissemination should be major tasks of present cancer research. Formation of metastases is favoured by both reduced numbers of immune cells in the bloodstream and impaired oxygen transport into tissues. These closely related signs often emerge concomitantly when the organism is endangered by circulating tumor cells released from the original tumor by therapeutic manipulations. From knowledge of these facts the O2-multistep immune-stimulation technique has been developed as a way of diminishing the risk of tumor spread. The process combines temporary elevation of the number of circulating immune cells with continuous improvement of oxygen transport into tissues.”
When the oxygen saturation of blood falls, conditions then become ripe for the creation of cancer. Oxygen is exchanged and removed from the arterial blood as it passes through the capillary system. If arterial blood is deficient in oxygen or if blocked arteries restrict the blood flow, then tissues oxygenated by the latter stages of the capillary system may be so deprived of oxygen as to become cancerous.
People with various degenerative diseases are often found to have low venous oxygen saturation. Once they receive proper treatment, the venous oxygen saturation level rises and their health and vitality improve dramatically.
Arterial oxygen saturation should ideally be very high. “High O2 tensions were lethal to cancer tissue, 95% being very toxic, whereas in general, normal tissue were not harmed by high oxygen tensions. Indeed, some tissues were found to require high O2 tensions”, J. B. Kizer quoted in “O2Xygen Therapies: A New Way of Approaching Disease” by McCabe, page 82.
He discovered a “switch mechanism” of blood microcirculation, which depends on the oxygen state of the body. A high value of pO2 (greater than or equal to 50 mm of Hg) at the venous ends of the capillaries, attainable by the procedures of the Oxygen Multistep Therapy and by powerful physical exercise as well, results in an increase of the blood microcirculation and, consequently, in a permanent elevation of the oxygen influx and uptake, respectively.
Anti-Inflammatory Oxygen Therapy increases the blood microcirculation and consequently we see a permanent elevation of the oxygen influx and uptake.
If the oxygen state gets worse and declines below a certain threshold, e.g. in progressing age or after long-term distress, the cross sections of the capillaries shrink by swelling of the endothelial cells, and the blood microcirculation will be diminished for an extended period. Reversing this degradation is quite a medical feat.
The utilization of the above-mentioned switch mechanism for permanent improvement of the oxygen flux into all the tissues of the organism is of decisive importance for fighting against the common cause of many diseases, disorders and complaints often going along with increasing age due to an insufficient oxygen (energy) supply for general metabolism.
On Professor von Ardenne’s site they say that, “This switching mechanism is interpreted as a re-enlargement of the capillary narrowed by oxygen deficiency(old age, disease, distress). The re-enlargement appears after increased oxygen uptake of the blood and improved oxygen utilization of human tissue over a certain time period.”
Resolved inflammation restores the blood supply to tissue – and allows the tissue to return to normal aerobic metabolism. Professor Ardenne showed that stress triggers persistent inflammation,which locks an escalating percentage of the body, and muscles into anaerobic metabolism – especially with advancing age.[2]
Anti-Inflammatory Oxygen Therapy specifically targets capillary inflammation with bursts of plasma dissolved oxygen at five times the level that were possible under the original design of Dr. von Ardenne.
Special Note: Dr. von Ardenne used magnesium for many reasons. When one looks at magnesium’s anti-inflammatory effect as well as its ability to increase oxygen carrying capacity we see why Ardenne was so insistent on magnesium. I became famous in the world of medicine after writing my Transdermal Magnesium Therapy book and I still use magnesium oil on a daily basis. Most do not take magnesium seriously when confronting cancer and other serious diseases. Researchers from Japan’s National Cancer Center in Tokyo have found that an increased intake of magnesium reduces a man’s risk of colon cancer by over 50 percent. Men with the highest average intakes of magnesium (at least 327 mg/d) were associated with a 52 percent lower risk of colon cancer, compared to men who consumed the lowest average intakes. Published in the Journal of Nutrition,[3] the research studied 87,117 people with an average age of 57 and followed them for about eight years. Dietary intakes were assessed using a food frequency questionnaire. Average intakes of magnesium for men and women were 284 and 279 milligrams per day.
Magnesium stabilizes ATP[4] allowing DNA and RNA transcriptions and repairs.[5] Magnesium repletion produces rapid disappearance of the periosteal tumors.[6]
Magnesium deficiency is carcinogenic, and in the case of solid tumors, a high level of supplemented magnesium inhibits carcinogenesis.[7] Both carcinogenesis and magnesium deficiency increase the plasma membrane permeability and fluidity.[8]
It has been suggested that magnesium deficiency may trigger carcinogenesis by increasing membrane permeability.[9] The membranes of magnesium-deficient cells seem to have a smoother surface than normal and decreased membrane viscosity, analogous to changes in human leukemia cells.[10],[11] There is drastic change in ionic flux from the outer and inner cell membranes (higher Ca and Na, lower Mg and K levels) both in the impaired membranes of cancer and of magnesium deficiency. In addition, we find that lead (Pb) salts are more leukemogenic when given to magnesium-deficient rats than when they are given to magnesium-adequate rats, suggesting that magnesium is protective.[12]
The School of Public Health at the Kaohsiung Medical College in Taiwan found that magnesium also exerts a protective effect against gastric cancer, but only for the group with the highest levels.[13]
Dr. Mark Sircus AC., OMD, DM (P)
References (13)
Fundamentals of combating cancer metastasis by oxygen multistep immunostimulation processes. von Ardenne M.; Med Hypotheses. 1985 May;17(1):47-65; http://www.ncbi.nlm.nih.gov/pubmed/3892251?ordinalpos=26&itool=EntrezSystem2…
Measurements and combat of stress effects (author’s transl); von Ardenne M.; ZFA. 1981;36(6):473-87; http://www.ncbi.nlm.nih.gov/pubmed/7336784
“High dietary intake of magnesium may decrease risk of colorectal cancer in Japanese men” Volume 140, Pages 779-785 Authors: E. Ma, S. Sasazuki, M. Inoue, M. Iwasaki, N. Sawada, R. Takachi, S. Tsugane, Japan Public Health Center-based Prospective Study Group.
Mg2+ is critical for all of the energetics of the cells because it is absolutely required that Mg2+ be bound (chelated) by ATP (adenosine triphosphate), the central high-energy compound of the body. ATP without Mg2+ bound cannot create the energy normally used by specific enzymes of the body to make protein, DNA, RNA, transport sodium or potassium or calcium in and out of cells, nor to phosphorylate proteins in response to hormone signals, etc. In fact, ATP without enough Mg2+ is non-functional and leads to cell death. Bound Mg2+ holds the triphosphate in the correct stereochemical position so that it can interact with ATP using enzymes and the Mg2+ also polarizes the phosphate backbone so that the ‘backside of the phosphorous’ is more positive and susceptible to attack by nucleophilic agents such as hydroxide ion or other negatively charged compounds. Bottom line, Mg2+ at critical concentrations is essential to life,” says Dr. Boyd Haley who asserts strongly that, “All detoxification mechanisms have as the bases of the energy required to remove a toxicant the need for Mg-ATP to drive the process. There is nothing done in the body that does not use energy and without Mg2+ this energy can neither be made nor used.” Detoxification of carcinogenic chemical poisons is essential for people want to avoid the ravages of cancer. The importance of magnesium in cancer prevention should not be underestimated.
Magnesium has a central regulatory role in the cell cycle including that of affecting transphorylation and DNA synthesis, has been proposed as the controller of cell growth, rather than calcium. It is postulated that Mg++ controls the timing of spindle and chromosome cycles by changes in intracellular concentration during the cell cycle. Magnesium levels fall as cells enlarge until they reach a level that allows for spindle formation. Mg influx then causes spindle breakdown and cell division.
Hunt, B.J., Belanger, L.F. Localized, multiform, sub-periosteal hyperplasia and generalized osteomyelosclerosis in magnesium-deficient rats. Calcif.Tiss.Res. 1972; 9:17-27.
Durlach J, Bara M, Guiet-Bara A, Collery P. Relationship between magnesium, cancer and carcinogenic or anticancer metals. Anticancer Res. 1986 Nov-Dec;6(6):1353-61.
Anghileri, L.J. Magnesium concentration variations during carcinogenesis.Magnesium Bull.1979; 1:46-48.
Blondell, J.W. The anticancer effect of magnesium.Medical Hypothesis 1980; 6:863-871.
Whitney, R.B., Sutherland, R.M. The influence of calcium, magnesium and cyclic adenosine 3’5’-monophosphate on the mixed lymphocyte reaction. J. Immunol. 1972; 108:1179-1183.
Petitou, M., Tuy, F., Rosenfeld, C., Mishal, Z., Paintrand, M., Jasmin, C., Mathe, G., Inbar, M. Decreased microviscosity of membrane lipids in leukemic cells; two possible mechanisms.Proc. Natl. Acad. Sci. USA 1978; 75:2306-2310.
Hass, G.M., McCreary, P.A., Laing, G.H., Galt, R.M. Lymphoproliferative and immumunologic aspects of magnesium deficiency. In Magnesium in Health and Disease (from 2nd Intl Mg Sympos, Montreal, Canada, 1976), b Eds. M. Cantin, M.S. Seelig, Publ. Spectrum Press, NY, 1980, pp 185-200.
Yang CY et al. Jpn J Cancer Res.1998 Feb;89 (2):124-30. Calcium, magnesium, and nitrate in drinking water and gastric cancer mortality.
Related Post
Oxygen, Baking Soda, and Magnesium Cure with Dr. Mark Sircus
Exercise with Oxygen Therapy (EWOT)
11 Facts About Oxygen You Probably Didn’t Know
How the Body Uses Oxygen
from WordPress https://ift.tt/2zwzffu via IFTTT
0 notes
Text
Find Doctors Who Treat MTHFR Symptoms at MTHFRdoctors.com
MTHFR is an enzyme that adds a methyl group to folic acid to make it usable by the body. The MTHFR gene produces this enzyme that is necessary for properly using vitamin B9. This enzyme is also important for converting homocysteine into methionine, which the body needs for proper metabolism and muscle growth and which is needed for glutathione creation. The process of methylation also involves the enzyme from the MTHFR gene, so those with a mutation may have trouble effectively eliminating toxins from the body.
Now for MTHFR Mutation you can find a Practitioner, MTHFR Doctors who treats MTHFR at MTHFRdoctors.com. MTHFR is a very important gene and its mutations may affect your health in many different ways. MTHFRdoctors.com experts have gathered a list of 10 important steps to help you learn what to do.
MTHFRdoctors.com offers to find MTHFR doctors, search for MTHFR practitioners who offer treatment for MTHFR mutation, MTHFR gene mutation treatment, MTHFR cancer treatment. The MTHFR practitioners offer help syndrome genetic, MTHFR epigenetics, MTHFR DNA and more.
The MTHFR gene test provides instructions for making an enzyme called methylenetetrahydrofolate reductase. This enzyme plays a role in processing amino acids, the building blocks of proteins. Methylenetetrahydrofolate reductase is important for a chemical reaction involving forms of the vitamin folate. Precisely, this enzyme transforms a molecule called 5, 10-methylenetetrahydrofolate to a molecule called 5-methyltetrahydrofolate. This reaction is required for the multistep process that converts the amino acid homocysteine to another amino acid, methionine. The body uses methionine to make proteins and other important compounds.
Individuals with a MTHFR gene mutation and MTHFR Symptoms have a highly reduced ability to convert folic acid or even folate into a usable form. There are many variations of the mutation, depending on how the gene was passed down from the parents. Individuals with low activity of the MTHFR enzyme may present with elevated homocysteine levels, which have been associated with inflammation and heart disease, birth defects, difficult pregnancies, and potentially an impaired ability to detoxify.
About MTHFRdoctors.com: As a leading online resource for everything related to MTHFR, MTHFRdoctors.com you will find Doctors, physicians and practitioners that are trained in treatments for MTHFR, Methylation Genetics, DNA, MTHFR test, COMT test and Genetic health reports. You can find information related to MTHFR Gene Mutation, Test Kits, Treatments and Health Tips at https://www.mthfrdoctors.com
0 notes
Text
Former National Cancer Institute and National Institutes of Health Scientist Calls Cancer “An Induced Disease of the Twentieth Century”


by Paul Fassa Health Impact News
Health Impact News first introduced the iconoclastic work of cellular biologist, Dr. Mahin Khatami, PhD, in an earlier article entitled HPV Vaccine Scam: NIH Scientist Exposes Corruption in Cancer and Vaccine Industries, which mentioned Dr. Khatami's extensive credentials that put her beyond criticism, though not beyond ignoring.
The pharmaceutical industry-controlled medical profession won't allow any light to shine on her 2016 white paper Safety concerns and hidden agenda behind HPV vaccines: another generation of drug-dependent society?
It, along with Dr. Khatami's impressive background, can be accessed here.
Now, Dr. Khatami, a former program director and health scientist administrator at the National Cancer Institute (NCI) and the National Institutes of Health (NIH), has published a new white paper exposing the fraud in the cancer industry, claiming that cancer is “an induced disease of the twentieth century.”
Dr. Khatami's Report That Shatters the Mythology of Orthodox Oncology
Dr. Khatami's 2018 white paper is titled Cancer; an induced disease of twentieth century! Induction of tolerance, increased entropy and 'Dark Energy': loss of biorhythms (Anabolism v. Catabolism). The phrase “induced disease” is an essential aspect of her critique on the cancer industry, as she tends to use the phrase “cancer establishment.”
Dr. Khatami does not avoid confronting the obvious regarding the current state of mainstream oncology. Unlike her somewhat less knowledgeable colleagues who avoid the obvious, Dr. Khatami has the biological molecular biochemical expertise to expertly put all the pieces together and allow the forest to appear despite conventional oncology's focus on the wrong trees.
Under Dr. Khatami's 2018 white paper section titled “Accidental discoveries in 1980s: time course kinetics of inflammation-induced developmental phases of immune dysfunction toward multistep tumorigenesis and angiogenesis” appear the following excerpts:
[Khatami's earlier] challenging efforts to promote the important role of inflammation in cancer research, diagnosis and therapy initially met with serious oppositions and denials by members of the establishment who rejected the submitted concepts and comprehensive proposals that were an extension of author's original discoveries.
However, the outcomes of reductionist approaches on identifications of hundreds of molecular entities that are used for drug development and claimed as 'targeted' therapy or 'personalized' or 'precision' medicine have been very disappointing, extremely costly and dangerous for patients and society. Majority of expensive projects in cancer research and therapy focus on identification of too many defective molecular species in the landscape of cancer molecular tsunami with little/no efforts to understand what initiate altered immune response profiles that lead to multistep tumorigenesis.
Later in her paper, Dr. Khatami goes to point out that vaccines and the multitude of environmental toxins introduced over the past six decades are mostly responsible for the proliferation of most diseases, including cancer, despite the “unlucky” random genetic mishap theories offered by some.
…decision makers in cancer establishment has gradually weakened and manipulated the autonomic biological circadian rhythms, the Yin and Yang of effective immunity, by introducing the public to various pathogen-specific vaccines and ingredients, in addition to exposures to exposures to a wide range of environmental hazards and low level carcinogens that made young and old in America sick and drug-dependent.
Analyses of related data suggest that nearly all diseases, to varying degrees, are the results of altered effective immunity or loss of biological circadian rhythms (on-off switches) that adversely influence tissue bioenergetics (mitochondria), the principal generator of energy and electricity (proton pumping) in organ systems.
Dr. Khatami explains the essence of her and her colleagues' earlier studies were demonstrating that the way to defeat cancer is to further explore how to influence immunity, by synchronizing metabolic biorhythms and regulating inflammation, as the better approach to protecting against and healing cancer.
This approach goes against the grain of conventional oncology's focus on each individual tree in the forest to forget the forest even exists. Current attempts at “killing” hundreds of arbitrarily contrived cancer types to involve many chemotherapeutic creations are at a 95 percent failure rate, Dr. Khatami states in this paper.
But, she adds, they enrich oncologists and the pharmaceutical industry, give work to researchers, and raise money to defeat cancer, instead of discovering ways to prevent or halt the “cancer tsunami.”
Dr. Jason Fung, MD, a fasting autophagy expert, agrees with Dr. Khatami's arguments against the widely held somatic mutation theory (SMT), which depicts the origin of cancer as simply a random collection of spontaneous genetic mutations rather than dietary and environmental epigenetic influences. He puts the current war on cancer's failure this way:
Despite the world of technology moving on a MagLev bullet train, and the world of medicine moving at a crawl, cancer remains standing still. This, despite billions of research dollars every year, more 'walks for cancer' than you can count, more pink ribbons, more tear-jerker stories on the mass media. Nobody wants to hear the truth, but here it is. The progress on cancer sucks. It really, really sucks. (Source)
Below is a graphic cycle of damaged immunity leading to the origination of a cancer tsunami:
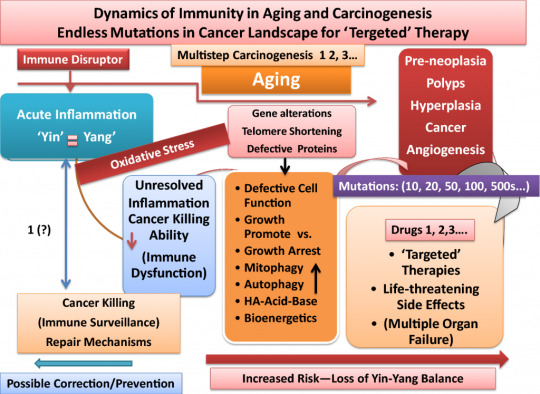
Throughout the rest of her paper, there are several sections that outline specific metabolic functions and dysfunctions that contribute to the origin of cancer with meticulously detailed scientific data, terminology, and other graphic illustrations.
Dr. Khatami summarizes her lengthy paper, containing 267 published study references, with:
[Earlier and more recent discoveries] suggest that cancer is only one disease. Disorderly growth of cancer cells is the results of loss of autonomic and synchronized balance in tumoricidal (Yin) and tumorigenic (Yang) properties of immunity to arrest cancer cells. Loss of differential bioenergetics in Yin (high energy, tumoricidal) and Yang (low energy, tumorigenic) pathways often leads to 'mild', 'moderate' or 'severe' immune disorders or cancer.
She concludes by explaining her paper as a roadmap that would hopefully lead to further research in the right direction of the various metabolic and immune responses, such as inflammation, that could be clinically influenced. Of course, this is all meant for the wayward cancer industry or establishment.
You can access Dr. Khatami's complete paper here.
Commentary
It's great that a renowned scientist with a high amount of establishment credentials is pointing out the ineffective, yet lucrative, fallacies and foibles of the cancer industry and is attempting to wake up said industry to diminish the amount of wasted money and destroyed lives caused by their expensive, toxic treatments.
Despite all the “sound and fury, signifying nothing” from and for the cancer industry, there are and have been, for decades longer than the “war on cancer,” several cancer cures discovered, created, and clinically-applied with success rates at/or above 80 percent, without side effects, and all without toxic chemical inventions.
They are not supported with governmental or private non-profit foundation funding, which mainstream oncology receives billions of dollars worth annually. They are not referred to by orthodox oncologists, even though those oncologists' attempts are failing. Many times MDs continue to gain financially from administering their agonizingly harmful procedures, even while their patients are dying.
The cancer industry denies the existence of “alternative” safe and successful solutions. They are not even noticed, except for the purpose of isolating them, attacking them, and scaring cancer victims away from them or shutting them down.
It's wiser to keep up with the latest on cancer prevention and alternative cures instead of waiting for those whose lucrative careers depend on not changing their paths to change their minds.
See also:
Health Impact News Article Archive of Cancer Prevention
Health Impact News Article Archive of Alternative Cancer Cures
The following video of Dr. Mahin Khatami's presentation is from the 4th Clinical Microbiology and Microbial Genomics Conference October 2015 in Philadelphia, PA.
youtube
<!--//<![CDATA[ var m3_u = (location.protocol=='https:'?'https://network.sophiamedia.com/openx/www/delivery/ajs.php':'http://network.sophiamedia.com/openx/www/delivery/ajs.php'); var m3_r = Math.floor(Math.random()*99999999999); if (!document.MAX_used) document.MAX_used = ','; document.write ("<scr"+"ipt type='text/javascript' src='"+m3_u); document.write ("?zoneid=3&target=_blank"); document.write ('&cb=' + m3_r); if (document.MAX_used != ',') document.write ("&exclude=" + document.MAX_used); document.write (document.charset ? '&charset='+document.charset : (document.characterSet ? '&charset='+document.characterSet : '')); document.write ("&loc=" + escape(window.location)); if (document.referrer) document.write ("&referer=" + escape(document.referrer)); if (document.context) document.write ("&context=" + escape(document.context)); if (document.mmm_fo) document.write ("&mmm_fo=1"); document.write ("'><\/scr"+"ipt>"); //]]>-->

0 notes
Text
What Type of Folate Should You Take?
What Type of Folate Should You Take?
There is a lot of confusion about folate, folic acid, MTHFR gene mutation, and supplementation. I am going to break it down for you in simple terms. I want you to be confident that you are getting enough of the right kind of folate for your body.
Major Players (like in a play)
Folate – water soluble B vitamin also known as B9 or folacin. Folate must be changed to a metabolically active form called methylfolate for our body to use it.
Folate status – amount of usable folate in the body. This is influenced by folate metabolism or how fast and how easily the body can change folate to its active form of methylfolate.
MTHFR enzyme- used to change folate or folic acid into an active form that the body can use.
MTHFR gene – makes the MTHFR enzyme (because they are both MTHFR this can be confusing). Many people have a gene mutation where they do not make enough of the enzyme MTHFR. This affects the folate status in the body.
Folic Acid - synthetic form of folate found in fortified foods and vitamin supplements. Folic acid has no biological activity unless converted into folates.
Methylfolate- active form of folate which goes by several names.
How do we get folate?
Folates are normally found in a wide variety of foods and are commonly consumed through a diet of green leafy vegetables, sprouts, fruits, brewer’s yeast, and animal products such as milk and dairy products, egg yolk and liver.
Unfortunately, folates contained in foods are unstable and susceptible to oxidation; they rapidly lose activity during food processing, manufacturing and storage and have a bioavailability range of 25-50%, depending on the kind of food. Fresh leafy vegetables stored at room temperature may lose up to 70% of their folate activity within three days and cooking it in water can increase the loss to 95%.
Humans cannot make folate and because of its water soluble nature, the body only stores folate to a limited extent. For this reason folate represents a dietary requirement and must be consumed in the diet. Neither folic acid nor food folates are biologically active and need to be converted to the metabolically active form.
So, if you want to get adequate amounts of folate it is important to eat the above foods fresh daily or to supplement with an active form of methylfolate that your body can use.
It is a multistep process to change folate to a useable active form of methylfolate and the enzyme MTHFR is required for this process. Some individuals do not produce adequate or effective amount of MTHFR enzymes due to the gene mutation called MTHFR: a genetic mutation that inhibits the ability of the body to methylate/convert folate or folic acid from the food we eat into the metabolite we need, L-5-Methyltetrahydrofolate. This means some individuals cannot actually use folate or folic acid from any source. For these people, because of the MTHFR gene mutation, they have to get folate in the active form to be able to use folate.
The correct type of folate to take
According to Dr. Ben Lynch an MTHFR expert you want to make sure your active form of methylfolate is the L form of methylfolate, which is a biologically active form, and not the D form of methylfolate, which is not biologically active and is possibly harmful to the liver where it is stored.
Here are the L forms which are well absorbed.
· L-5-MTHF =
L-5-Methyltetrahydrofolate =
6(S)-L-MTHF =
6(S)-L-Methyltetrahydrofolate
· L-Methylfolate Calcium =
Metafolin =
Levomefolic Acid
Another good form is Quatrefolic which passes the gastric barrier and is absorbed mainly in the small intestine and has higher uptake of folate.
Here are the D forms which are chemically not biologically active forms and possibly harmful to the liver.
D-5-MTHF =
D-5-Methyltetrahydrofolate =
6(R)-L-MTHF =
6(R)-L-Methyltetrahydrofolate
These next 3 forms do not say they are L form or 6(S) form so may have more then 1% of the D form, which is hard for the body to use. They may or may not be biologically active methylfolate, so you either need to contact the company and ask or avoid them.
5-MTHF 5-Methylfolate 5-Methyltetrahydrofolate
There you go, the specifics on how to know which type of folate you should take. You can see the types of folate I recommend here.
Sources
Email from Dr. Katherine Gardner Yates, D.C. Chiropractor and expert on nutritional principles and natural healing philosophy.
http://mthfr.net/
Picture in this post are copyright under Creative Commons Zero (CC0) license
#folate in pregnancy#folic acid#mthfr#mthfr and pregnancy#midwife advice#prenatal vitamins#methylfoate#mthfr and miscarriage
0 notes
Text
Cancers, Vol. 10, Pages 191: WIP-YAP/TAZ as A New Pro-Oncogenic Pathway in Glioma
Cancers, Vol. 10, Pages 191: WIP-YAP/TAZ as A New Pro-Oncogenic Pathway in Glioma
Cancers doi: 10.3390/cancers10060191
Authors: Sergio Rivas Inés M. Antón Francisco Wandosell
Wild-type p53 (wtp53) is described as a tumour suppressor gene, and mutations in p53 occur in many human cancers. Indeed, in high-grade malignant glioma, numerous molecular genetics studies have established central roles of RTK-PI3K-PTEN and ARF-MDM2-p53 INK4a-RB pathways in promoting oncogenic capacity. Deregulation of these signalling pathways, among others, drives changes in the glial/stem cell state and environment that permit autonomous growth. The initially transformed cell may undergo subsequent modifications, acquiring a more complete tumour-initiating phenotype responsible for disease advancement to stages that are more aggressive. We recently established that the oncogenic activity of mutant p53 (mtp53) is driven by the actin cytoskeleton-associated protein WIP (WASP-interacting protein), correlated with tumour growth, and more importantly that both proteins are responsible for the tumour-initiating cell phenotype. We reported that WIP knockdown in mtp53-expressing glioblastoma greatly reduced proliferation and growth capacity of cancer stem cell (CSC)-like cells and decreased CSC-like markers, such as hyaluronic acid receptor (CD44), prominin-1 (CD133), yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ). We thus propose a new CSC signalling pathway downstream of mtp53 in which Akt regulates WIP and controls YAP/TAZ stability. WIP drives a mechanism that stimulates growth signals, promoting YAP/TAZ and β-catenin stability in a Hippo-independent fashion, which allows cells to coordinate processes such as proliferation, stemness and invasiveness, which are key factors in cancer progression. Based on this multistep tumourigenic model, it is tantalizing to propose that WIP inhibitors may be applied as an effective anti-cancer therapy.
https://ift.tt/2sNqj0O
0 notes
Text
Molecular pathogenesis and targeted therapies for NOTCH1-induced T-cell acute lymphoblastic leukemia
http://leukemiaandlymphoma.eu/leukemia-articles/molecular-pathogenesis-and-targeted-therapies-notch1-induced-t-cell-acute/fulltext
Molecular pathogenesis and targeted therapies for NOTCH1-induced T-cell acute lymphoblastic leukemia Maddalena Paganin a and Adolfo Ferrando a b c lowast Blood Reviews, 2, 25, pages 83 - 90
Abstract T-cell acute lymphoblastic leukemia (T-ALL) is an aggressive hematologic tumor resulting from the malignant transformation of immature T-cell progenitors. Originally associated with a dismal prognosis, the outcome of T-ALL patients has improved remarkably over the last two decades as a result of the introduction of intensified chemotherapy protocols. However, these treatments are associated with significant acute and long-term toxicities, and the treatment of patients presenting with primary resistant disease or those relapsing after a transient response remains challenging. T-ALL is a genetically heterogeneous disease in which numerous chromosomal and genetic alterations cooperate to promote the aberrant proliferation and survival of leukemic lymphoblasts. However, the identification of activating mutations in the NOTCH1 gene in over 50% of T-ALL cases has come to define aberrant NOTCH signaling as a central player in this disease. Therefore, the NOTCH pathway represents an important potential therapeutic target. In this review, we will update our current understanding of the molecular basis of T-ALL, with a particular focus on the role of the NOTCH1 oncogene and the development of anti-NOTCH1 targeted therapies for the treatment of this disease. Keywords: T-ALL, NOTCH, Gamma-secretase inhibitors, Targeted therapy, Prognosis, Leukemia.
Introduction Acute lymphoblastic leukemias (ALL) are characterized by the uncontrolled clonal proliferation of immature lymphoid cells which infiltrate the bone marrow. In T-cell acute lymphoblastic leukemias (T-ALL) the malignant clone is derived from T-cell progenitor cells and expresses immature immunophenotypic markers characteristic of the T-cell lineage. T-ALL represents about 15% of pediatric and 25% of adult ALLs and is typically associated with very high white cell counts, mediastinal masses with pleural effusions, and increased risk of leptomeningeal infiltration at diagnosis. 1 Although initially associated with a particularly bad prognosis, the introduction of intensified treatment protocols has improved the outcome of this disease and current therapies achieve five-year relapse-free survival rates of about 75% in children and 50% in adults.2, 3, 4, 5, 6, 7, and 8 T-cell transformation is a multistep oncogenic process in which multiple lesions involving different oncogenes and tumor suppressor genes cooperate to disrupt the normal circuitry that controls cell proliferation, differentiation and survival during T-cell development.9, 10, 11, and 12 Most of what we know about the molecular basis of T-ALL has been learned from the study of recurrent cytogenetic alterations. 9 The most common genetic alteration in T-ALL is the presence of deletions in the CDKN2A tumor suppressor locus containing the P16/INK4A and the P14/ARF tumor suppressor genes, which control cell cycle progression and p53 mediated apoptosis, respectively. 13 In addition, over 50% of T-ALLs harbor activating mutations in the NOTCH signaling pathway making of NOTCH1 the most prominent T-ALL specific oncogene 14 and defining T-ALL as a disease primarily characterized by aberrant NOTCH1 activation.15 and 16 However, T-ALL is a heterogeneous disease in which different molecular groups, primarily defined by the expression of T-ALL transcription factor oncogenes, are associated with specific patterns of gene expression, a specific block in cell differentiation and distinct clinical characteristics.10, 11, and 12 Thus, T-ALL-associated chromosomal translocations typically result in the juxtaposition of a selective group of oncogenic transcription factors next to strong regulatory elements located in the vicinity of the T-cell receptor β (TCRB) gene in chromosome 7q34 or the T-cell receptor α-δ (TCRAD) locus in chromosome 14q11.9 and 17 These T-ALL-specific transcription factor oncogenes include basic helix–loop–helix (bHLH) family members such as TAL1,18, 19, 20, and 21TAL2, 22 LYL1 23 and BHLHB1 24 ; LIM-only domain (LMO) factors such as LMO1 and LMO225, 26, 27, 28, and 29; TLX1/HOX11,30, 31, 32, and 33TLX3/HOX11L2, 34 NKX2.535 and 36 and HOXA homeobox genes11 and 37; MYC38, 39, 40, 41, and 42 and MYB 43 oncogenes; and TAN1, a truncated and constitutively activated form of the NOTCH1 receptor.44 and 45 In some cases, these factors can also be activated in the context of other non-TCR-associated chromosomal abnormalities. This is the case for small deletions activating TAL1 46 and LMO2 47 ; duplications of the MYB oncogene48 and 49; and the t(5;14)(q32;q11) translocation which activates the TLX3 oncogene in chromosome 5 by relocating it to the vicinity of the BCL11B locus in chromosome 14. Additional molecular alterations present in T-ALL include transcription factor fusion oncogenes such as PICALM/MLLT10/CALM-AF10,50, 51, and 52MLL-MLLT1/MLL-ENL,53 and 54SET/NUP214, 55 NUP98-RAP1GDS156 and 57; activation of signaling factors driving proliferation such as LCK, 58 CCND2,59 and 60JAK1, 61 NUP214-ABL1, 62 EML1-ABL1, 17 and NRAS63 and 64; and the loss of tumor suppressor genes in the RAS (NF1 65 ) and PI3K (PTEN 66 ) signaling pathways. However, the catalog of genetic alterations involved in the pathogenesis of T-ALL is not yet complete as shown by the recent identification of loss-of-function mutations in WT1, 67 the PTPN2 phosphatase 68 and the PHF6 tumor suppressor gene. 69
NOTCH1 signaling pathway The NOTCH1 receptor is a class I transmembrane protein that functions as a ligand-activated transcription factor ( Fig. 1 ). 15 Thus, NOTCH1 directly transduces information from extracellular signals into changes in gene expression in the nucleus. The main components of NOTCH1 signaling include: the Delta/Serrate/LAG-2 (DSL) family of ligands (Delta-like 1, 3, and 4 as well as Jagged 1 and 2); the NOTCH1 receptor (NOTCH1); the RBPJ/CSL (CBF1/Su(H)/LAG-1) DNA-binding protein; and the mastermind-like family of coactivators. In resting conditions, NOTCH1 sits in the membrane as a heterodimeric complex composed of an N-terminal extracellular subunit (NEC) and a C-terminal transmembrane and intracellular subunit (NTM). The NEC subunit interacts with Delta-like and Jagged ligands through 36 epidermal growth factor (EGF)-like repeat domains. In addition, it contains a negative regulatory region (NRR) composed of three Lin12/NOTCH repeats (LNRs). These LNR domains fold over and stabilize the heterodimerization domain (HD), which consists of the C-terminus of NEC and the N-terminus of NTM in close interaction, to prevent the spontaneous activation of the receptor in the absence of ligand. The NTM subunit contains a transmembrane sequence followed by a series of cytoplasmic domains, including a RAM domain, a series of ankyrin repeats, a transactivator domain, and several nuclear localization signals, which collectively function as a ligand-activated transcription factor. The NTM also contains a C-terminal PEST (proline (P), glutamic acid (E), serine (S), and threonine (T) rich) domain, which is responsible for the proteosomal degradation of activated NOTCH1 in the nucleus. 15 gr1 Fig. 1 NOTCH1 mutations in T-ALL. Schematic representation of the NOTCH1 receptor structure and types of NOTCH1 mutations found in T-ALL. EGF-like, EGF-like repeats involved in ligand recognition. NRR, negative regulatory region. LNR, Lin12/NOTCH repeats. HD, heterodimerization domain. RAM, RAM domain. ANK, ankyrin repeats. TAD, transactivation domain. PEST, proline, glutamic acid, serine and threonine rich domain. ICN1, intracellular NOTCH1. Under physiologic conditions the NOTCH1 receptor is activated via interaction with a Jagged or Delta-like ligand molecule. This ligand–receptor interaction induces a conformational change in the NRR regulatory region and triggers the cleavage of the HD domain by the ADAM10 and ADAM17 metalloproteases at the cell surface.70, 71, 72, 73, and 74 This first activation-associated cleavage, also known as S2, is then followed by a second proteolytic cleavage (S3) catalyzed by the γ-secretase complex in the transmembrane region of the receptor.70, 71, and 72 Thus, the γ-secretase complex releases the intracellular domain of NOTCH1 (ICN1) into the cytosol and allows its translocation into the nucleus, where it associates with the RBPJ/CSL DNA-binding protein, recruits members of the mastermind (MAML) family of coactivators and p300, and through these interactions, activates gene expression. 15 Finally, recruitment of the RNA polymerase II holoenzyme to the ICN1-RBPJ/CSL-MAML transcriptional complex triggers the phosphorylation of the PEST domain of NOTCH1 by cyclin-dependent kinase 8 and recruits the FBXW7/SCF ubiquitin ligase complex, which ultimately mediates the polyubiquitination and proteasomal degradation of the activated receptor in the nucleus. 15
NOTCH1 in T-cell development The NOTCH signaling pathway is responsible for cell fate specification and tissue patterning in multiple cellular and tissue contexts during development. In the lymphoid system NOTCH signals provided by the thymic microenvironment are essential for the specification and development of T-cell progenitors.75 and 76 Consistent with this model, conditional inactivation of NOTCH1 results in a complete ablation of T-cell lymphopoiesis and differentiation accompanied by ectopic B-cell development in the thymus.77 and 78 Likewise, overexpression of an active, intracellular form of NOTCH1 in bone marrow progenitors results in ectopic pre-T-cell development in the bone marrow. 79 Upon T-cell specification, thymocytes differentiate into αβ or γδ T-cell lineages, and while the development of γδ T-cells seems to be independent of NOTCH,80 and 81 αβ T-cells require continuous NOTCH1 activation for their maturation to the DN3 stage of development. 81 During this process, several important factors required for T-cell development are transcriptionally controlled by NOTCH1, including the pre-T-cell receptor alpha (PTCRA), 82 the IL7 receptor alpha (IL7RA) 83 and MYC. 84 Both preTCR signaling and NOTCH activation are needed for growth and survival at the so called β-selection checkpoint, 80 at which point NOTCH1 signaling is critically required to sustain cell metabolism via activation of the PI3K–AKT cascade. 85
Aberrant NOTCH1 activation in T-ALL The first evidence of the role of NOTCH1 in the pathogenesis of T-ALL resulted from the cloning of TAN1, a truncated and constitutively active form of NOTCH1, at the breakpoint of the t(7;9)(q34;q34.3) chromosomal translocation present in about 1% of T-ALL cases. 44 In this translocation, the NOTCH1 locus in 9q34 is broken so that the derivative chromosome 9 retains the N-terminal domains of NOTCH1, including the NRR region, while the transmembrane and intracellular domains of the receptor are translocated to the derivative chromosome 7 where they are aberrantly expressed under the control of the TCRB regulatory sequences. Ultimately this rearrangement results in constitutive activation of NOTCH1 signaling due to the expression of high levels of NTM and/or ICN1 in T-cell precursors ( Fig. 1 ).44 and 45 The pathogenic role of activated NOTCH1 in T-ALL was fully demonstrated when irradiated mice reconstituted with bone marrow progenitors expressing activated forms of NOTCH1 developed clonal hematopoietic tumors characterized as T-ALL. 86 In addition, T-cell tumors generated by insertional mutagenesis showed a high incidence of retroviral integrations resulting in constitutive activation of NOTCH1. 87 However, it was only after the identification of activating mutations in NOTCH1 in over 50% of human T-ALL cases ( Fig. 1 ) 14 that the central role of NOTCH1 in the pathogenesis of this disease was fully appreciated. Activating mutations in NOTCH1 typically result in the disruption of molecular locks responsible for preventing the spontaneous activation of the receptor at the membrane or mediating the termination of NOTCH1 signaling in the nucleus. 14 Thus, most mutations in the HD domain (exon 26 and exon 27), which are present in approximately 40% of human T-ALLs, destabilize the interaction between the N-terminal and C-terminal HD subunits and result in ligand-independent activation or ligand hypersensitivity (HD class 1 mutations) ( Fig. 1 ). 88 A second mutational hotspot is located at the 3′ end of the gene, which encodes the C-terminal PEST domain. 14 PEST domain mutations are present in about 15% of T-ALL samples and are typically truncating and nonsense mutations, which result in deletion of the recognition sequence for proteasomal degradation of ICN1 by the FBXW7/SCF complex in the nucleus ( Fig. 1 ). In rare cases, NOTCH1 is activated as the result of in-frame insertions in the distal part of the HD domain (HD class 2 mutations) that result in the displacement and constitutive processing at the ADAM protease cleavage site. 88 Alternative mechanisms of NOTCH1 activation include juxtamembrane expansion NOTCH1 mutations (JME alleles), which consist of extracellular in-frame insertions that displace the HD domain away from the membrane ( Fig. 1 ) 89 and the NOTCH1 H1545P mutation—located in the NOTCH1 LNR-C repeat—that disrupts the activity of the NRR and facilitates S2 processing of the HD domain ( Fig. 1 ). 90 In addition to these mutations in the NOTCH1 gene, about 15% of T-ALLs harbor mutations in FBXW7, which typically involve key arginine residues responsible for the recognition of phosphorylation sites in the PEST domain of NOTCH1.91, 92, and 93 These FBXW7 mutations impair the substrate recognition function of the FBXW7/SCF complex and impair the degradation of activated NOTCH1 ( Fig. 1 ).91 and 92 In addition, the oncogenic effects of FBXW7 mutations may extend beyond the NOTCH1 signaling pathway as this ubiquitin ligase also mediates the proteosomal degradation of additional oncoproteins such as MYC, JUN, Cyclin E, Aurora-A and mTOR.94, 95, and 96 Finally, about 20% of T-ALL patients harbor either dual mutations in the HD and PEST domains of NOTCH1 or both a NOTCH1 HD allele and a FBXW7 mutation. The combined effect of these mutations results in exceedingly high levels of NOTCH signaling as a result of NOTCH ligand-independent activation at the membrane plus impaired ICN1 degradation in the nucleus.14 and 91 An important point worth emphasizing here is that not all NOTCH mutations are functionally equivalent. Indeed each of the different types of NOTCH1 alleles described above has very different effects in its mechanism of action and its level of activation. NOTCH1 PEST mutations, when present alone, are typically weak alleles and are predicted to be functional only in the presence of NOTCH ligands. 14 NOTCH1 HD alleles result in variable levels of spontaneous NOTCH1 activation and, although some may induce ligand-independent activation of the receptor, others probably only confer ligand hypersensitivity. 88 In contrast, truncated NOTCH1 alleles resulting from the t(7;9) translocation, NOTCH1 insertion mutations (class 2 HD mutations and JME alleles), and double mutant alleles (NOTCH1 HD plus PEST or NOTCH1 HD plus FBXW7 mutations) result in remarkably high levels of NOTCH1 activation. 14 Consistently, each of these alleles and allele combinations has shown very different effects in its capacity to induce leukemia when expressed in mouse hematopoietic progenitor cells. 97 Specifically, weak NOTCH1 alleles failed to induce T-ALL by themselves although they accelerate T-cell transformation in hematopoietic progenitors expressing the k-ras oncogene. 97 Overall, strong NOTCH1 mutants may work as major drivers of the tumor phenotype, acting potentially probably as initiating events in T-ALL, while weaker alleles may function as secondary events that contribute to tumor progression.
Genes and pathways controlled by NOTCH1 in T-cell transformation The identification of genes and pathways controlled by NOTCH in T-ALL has been the focus of extensive research over the last years. These studies have defined a prominent role for NOTCH1 as a central regulator, promoting leukemia cell growth by multiple direct and indirect mechanisms. Gene expression profiling of T-ALL cell lines and ChIP-on-chip analysis of NOTCH1 in T-ALL cells revealed a prominent role of oncogenic NOTCH1 as a direct transcriptional activator of multiple genes involved in anabolic cell growth and metabolism. 98 In addition, this study also identified the MYC oncogene as a prominent direct target gene regulated by NOTCH1 in human leukemias. 98 Notably, most of the genes controlled by NOTCH1 that regulate cell growth, proliferation and metabolism are also targets of MYC.98 and 99 The resulting NOTCH1–MYC feed-forward-loop transcriptional regulatory network reinforces the expression of genes implicated in anabolic pathways, ribosome biosynthesis, protein translation and nucleotide and amino acid metabolism downstream of NOTCH1.84 and 98 Consistent with these observations, analysis of mouse tumor cells also revealed c-Myc as a prominent NOTCH1 target gene in T-cell transformation.84 and 100 In addition to its direct effect on anabolic genes and facilitating cell growth via upregulation of MYC, NOTCH1 facilitates the activation of the PI3K–AKT–mTOR signaling pathway, a critical regulator of cell growth and metabolism.66 and 85 The first indication of the key interaction between NOTCH and the PI3K pathway was provided in a seminal manuscript by Ciofani and coworkers who demonstrated that NOTCH signals regulate cell size, glucose uptake and glycolysis via activation of the PI3K–AKT signaling pathway during T-cell development. 85 More recently, phosphoproteomic analysis demonstrated a marked suppression of mTOR signaling in T-ALL cells upon inhibition of NOTCH signaling. 101 Overall, NOTCH1 seems to facilitate the activation of the PI3K–AKT–mTOR pathway at multiple levels. In T-cell progenitors and T-ALL lymphoblasts, a transcriptional repressor directly downstream of NOTCH1 signaling, HES1, can downregulate the expression of PTEN, a critical negative regulator of the PI3K pathway. 66 In addition NOTCH1 can activate AKT via the LCK tyrosine kinase in T-cells 102 and MYC can rescue the inhibitory effects of blocking NOTCH1 on the mTOR pathway. 101 Finally, numerous signaling molecules upstream of PI3K, including the interleukin 7 receptor alpha chain (IL7RA) 83 and the pre-T-cell receptor alpha (PTCRA), 82 are upregulated upon activation of NOTCH1 signaling in T-cell progenitors and in T-ALL lymphoblasts. The transcriptional program activated by oncogenic NOTCH1 also has a direct effect on cell cycle progression. For instance, oncogenic NOTCH1 signaling promotes G1/S cell cycle progression in T-ALL.14, 45, 103, 104, and 105 These effects are mediated in part by transcriptional upregulation of CCND3, CDK4, and CDK6. 103 Notably, CCND3 is a direct NOTCH1 target gene in T-ALL and is strictly required for NOTCH1-induced transformation. 106 Moreover, inhibition of NOTCH signaling in T-ALL is associated with upregulation of the cyclin-dependent kinase inhibitors CDKN2D (P19/INK4d) and CDKN1B (p27/Kip1).98 and 105 Finally, in hematopietic progenitors, NOTCH1 can induce the transcription of the S phase kinase-associated protein 2 (SKP2), which mediates the proteasomal degradation of CDKN1B (p27/Kip1) and CDKN1A (p21/Cip1). 104 Finally, NOTCH1 signaling can also regulate the survival of T-ALL cells via interaction with the NFκB. Specifically, activation of NOTCH signaling upregulates NFκB activity by increasing expression of IkB-kinase 107 and upregulating both the expression and the nuclear localization of NFκB. 108 The critical role of this interaction is demonstrated by the antileukemic effects of NFκB inhibition in T-ALL and the strict requirement of NFκB signaling for NOTCH-induced transformation. 109
NOTCH1 mutations and clinical prognosis in T-ALL Since the identification of activating mutations in NOTCH1, a number of studies have addressed the prognostic significance of these alterations in T-ALL. Initially, a study reporting results from a cohort of 157 pediatric T-ALL patients treated with the ALL-BFM 2000 protocol found that NOTCH1 mutations were associated with increased prednisone sensitivity, lower levels of minimal residual disease and favorable long-term outcomes. 110 Similarly, analysis of 55 pediatric T-ALL and 14T-cell lymphoblastic lymphoma patients treated in the Japan Association of Childhood Leukemia Study (JACLS) protocols ALL-97 and NHL-98 showed an improved outcome in patients harboring NOTCH1 and/or FBXW7 mutations. 111 In adult T-ALL, analysis of patients treated in the LALA-94 or GRAALL-2003 studies also identified NOTCH1 and/or FBXW7 mutations as favorable prognostic markers. 112 However, these results have not been fully validated in other series. Analysis of 72 pediatric T-ALL patients treated with the ALL-7, ALL-8 or ALL-9 protocols by the Dutch Childhood Oncology Group 113 and a study analyzing a cohort of 88 adult T-ALL patients treated according to the MRC UKALLXII/ECOGE2993 protocol 114 failed to detect a significant association between NOTCH1 and FBXW7 mutations and clinical outcome. Notably, three timely reports have recently readdressed the association of NOTCH activation with outcome, clarifying some of the uncertainties raised by earlier studies. First, a retrospective study on the relevance of NOTCH1/FBXW7 mutations in pediatric T-ALL analyzed patients enrolled on Dutch DCOG ALL-7/8 or ALL-9 or the German COALL-97 protocols and combined mutation analysis of NOTCH1 and FBXW7 with direct measurement of activated NOTCH1 protein using reverse-phase protein microarrays. 115 This analysis confirmed that NOTCH1 and FBXW7 mutations are associated with increased intracellular NOTCH1 levels in clinical samples. 115 In this series, the presence of NOTCH1/FBXW7 mutations was associated with a good initial in vivo prednisone response. However, this improved response to therapy did not translate into a superior outcome. 115 Similarly, analysis of NOTCH1 and FBXW7 mutations in 134 children with T-ALL enrolled in EORTC-CLG trials showed that mutation-positive patients have a better response to prephase therapy and lower levels of minimal residual disease at the end of induction. 116 However, this improved therapeutic response once again, did not result in improved outcome. 116 Finally, and in contrast with the results of these reports, an extended analysis of the effects of NOTCH1 and FBXW7 mutations in patients treated on ALL-BFM protocols confirmed the overall favorable effect of activating NOTCH1 mutations in prognosis originally observed in the BFM2000 study. 117 This series included 151 cases from the original report of the ALL-BFM 2000 protocol 110 and extended this series by including 150 new cases. NOTCH1 and FBXW7 mutations in this cohort were associated with rapid early treatment response both in terms of prednisone sensitivity and as measured by minimal residual disease. 117 Notably, this improved therapeutic response resulted in improved outcome and decreased risk of relapse. 117 Overall the results of these studies show that activation of NOTCH pathway is associated with improved therapeutic response and high sensitivity to glucocorticoid therapy in T-ALL. However, the ultimate effect of these mutations in terms of clinical outcome seems to be therapy-dependent.
Targeted inhibition of NOTCH1 for the treatment of T-ALL Perhaps the most exciting opportunity derived from the identification of NOTCH1 mutations in T-ALL is the possibility of developing anti-NOTCH1 targeted therapies in this disease. The γ-secretase complex, responsible for the proteolytic processing and activation of NOTCH signaling can be inhibited with small molecule inhibitors (GSIs) and has been the focus of extensive research by pharmaceutical companies because of its role in the pathogenesis of Alzheimer's disease.118 and 119 These GSIs function as pan-NOTCH inhibitors blocking the activity of all 4 NOTCH receptors. Early studies on the activity of GSIs as an anti NOTCH-therapy for T-ALL showed that treatment of T-ALL cell lines with these drugs resulted in rapid clearance of activated NOTCH1 protein and effective downregulation of NOTCH1 target genes.14, 45, 84, 98, and 101 Most notably, NOTCH inhibition reduced growth and proliferation by inducing G1 cell cycle arrest and decreasing cell size.14, 45, 66, and 101 Following these encouraging results, the Dana-Farber Cancer Institute performed a phase I clinical trial testing MK-0752, an oral GSI developed by Merck for the treatment of Alzheimer's disease, in T-ALL patients. 120 Six adults and two children with leukemia (seven with T-ALL and one with AML) where enrolled in this study and four of the seven T-ALL patients showed activating mutations in NOTCH1. Treatment duration ranged from 2 to 56 days, and one patient with T-ALL and a NOTCH1 mutation achieved a 45% reduction in a mediastinal mass after 28 days. However, this patient subsequently progressed, and no patient achieved an objective response before discontinuation because of disease progression or drug-related toxicity. 120 The most common dose-limiting toxicity was grade 3/4 diarrhea, revealing an unfavorable toxicity profile most probably related to inhibition of NOTCH signaling in the gut. The development of gastrointestinal toxicity in the context of GSI therapy was not completely unanticipated and has emerged as a significant obstacle for the clinical development of these drugs. NOTCH1 and NOTCH2 play an important role in the intestinal epithelium, where they are involved in the control of cell proliferation and differentiation, and as noted above, GSIs are pan-NOTCH inhibitors that cause a systemic block of all 4 NOTCH receptors. Genetic inhibition of NOTCH signaling in the gut using animal models via deletion of the Rbpjk gene 121 or in the context of double Notch1/Notch2 conditional knockouts 122 induces cell cycle arrest and differentiation to secretory cell lineages at the expense of the absorptive epithelium; a phenotype that is recapitulated upon pharmacologic inhibition of the Notch pathway with GSIs.121 and 123 Overall, these results strongly suggest that alternative strategies with an improved therapeutic window may be needed for the successful implementation of GSIs as anti-NOTCH therapies in T-ALL. In this regard, a recent report from Merck has shown that three days of > 70% Notch inhibition with a GSI is sufficient to induce effective antileukemic responses in T-ALL xenograft models and is well-tolerated. 124 A similar intermittent dosing approach has shown to reduce the toxicity associated with PF-03084014, a GSI developed by Pfizer. 125 These results illustrate that secretory metaplasia induced by GSIs is time- and dose-dependent and can be avoided using intermittent dosing schemes. An alternative approach to improve the safety and efficacy of anti-NOTCH therapies in T-ALL may result from the combined used of GSIs with chemotherapy or other molecularly targeted drugs. The idea is to use GSIs at high doses for short periods of time to avoid the development of gastrointestinal toxicity while using drug combinations that increase their antileukemic efficacy. Combination therapies of GSIs with CDK inhibitors, 105 drugs targeting NFκB signaling, 91 or small molecule inhibitors of CK2 125 and the PI3K–AKT–mTOR pathway66, 101, and 126 have been shown to increase the antileukemic effects of these pan-NOTCH inhibitors. In addition, prolonged exposure to GSIs may increase the response to glucocorticoid treatment, 127 and inhibition of NOTCH signaling with a GSI can sensitize glucocorticoid-resistant T-ALL cell lines to glucocorticoid-induced apoptosis. 128 Importantly, in vivo testing of GSIs and glucocorticoids in combination in a mouse model of glucocorticoid-resistant T-ALL showed that glucocorticoid treatment has a direct protective effect against GSI-induced intestinal toxicity in mice.128 and 129 These results have now been confirmed and extended in a report showing that glucocorticoids abrogate the gastrointestinal toxicity induced by the GSI PF-03084014 and that delayed administration of glucocorticoids does not impair their protective effect against GSI-induced gut toxicity. 130 Overall, these results strongly suggest that glucocorticoid treatment may enhance the antileukemic effects of GSIs, while at the same time amelliorating the intestinal toxicity typically associated with systemic inhibition of NOTCH signaling. 128
Modulators of clinical response to GSI in T-ALL Despite the prominent role of NOTCH1 in the pathogenesis of T-ALL, inhibition of NOTCH1 signaling seems to have only modest antileukemic effects against human T-ALL cell lines. Thus, inhibition of NOTCH signaling with GSIs is effective only in a fraction of these tumors and induces primarily a cytostatic effect,14, 45, and 98 although it can also result in the induction of apoptosis in some instances.45, 124, and 130 In contrast, Notch-induced mouse T-ALLs seem to be more sensitive to inhibition of NOTCH signaling.97 and 126 Comparative analysis of GSI-sensitive and GSI-resistant T-ALL cell lines showed that GSI treatment can effectively decrease the level of active NOTCH1 protein and the expression of NOTCH1 target genes in both sensitive and resistant tumors.66 and 98 These results demonstrate that GSI resistance in T-ALL cell lines is not mediated by defects in drug uptake or impaired inhibition of the γ-secretase complex and suggests that human T-cell leukemia cell lines may have accumulated additional mutations that sustain leukemic cell growth and bypass the effects of NOTCH1 inhibition. Detailed molecular analysis of GSI-sensitive and GSI-resistant T-ALL cell lines showed a striking correlation between PTEN mutational status and GSI sensitivity, as all GSI-sensitive tumors were PTEN wild type while each of the GSI-resistant lines analyzed showed mutational loss of this tumor suppressor gene. 66 However, analyses of mouse models of NOTCH1-induced leukemias and primary T-ALL samples in culture suggest that additional mutations may be required to confer full resistance to GSI therapy. 131 Notably, FBXW7 mutations, which upregulate the expression of MYC, JUN and Cyclin E in addition to contributing to increased ICN1 stability, are also more prevalent in GSI-resistant T-ALL cell lines.91 and 92 As clinical trials testing the safety and efficacy of GSI therapy in T-ALL progress, it will be important to analyze the effect of these genetic alterations in the response to anti-NOTCH1 therapies.
New and emerging anti-NOTCH therapies The limitations of GSIs in the clinic suggest that alternative strategies may be needed for the therapeutic targeting of NOTCH1 in T-ALL. One possibility resides in the use of synthetic peptides to block the NOTCH transcriptional complex directly in the cell nucleus. This approach would confer direct NOTCH inhibition and may have a more rapid inhibitory effect on NOTCH signaling in the cell than GSIs. Following this approach, Moellering and coworkers have recently shown that SAHM1, a cell-permeable, stabilized alpha-helical peptide targeting the protein–protein interface responsible for the recruitment of MAML1 into the NOTCH–CSL transactivation complex, can effectively block NOTCH signaling and has potent, NOTCH-specific antileukemic effects both in human T-ALL cell lines and in a mouse model of NOTCH1-induced T-ALL. 132 Finally, given that NOTCH proteins are surface molecules, specific antibodies could provide selective blocking of NOTCH1, specifically, while preserving the activity of the other three NOTCH family members. An elegant study by Wu and coworkers at Genentech has demonstrated that highly specialized antibodies can block NOTCH1 signaling by binding to and stabilizing the LNR–HD complex, 133 locking the receptor in an “off” conformation. Notably, this anti-NOTCH1 antibody blocked leukemic cell growth in pre-clinical models and inhibited angiogenesis. 133 Moreover, the anti-Notch1 antibody did not affect the activity of Notch2, which precluded the development of overt gastrointestinal toxicity. 133 In a related study, Aste-Amézaga and coworkers showed that anti-NOTCH1 antibodies can block ligand-independent signaling driven by Notch1 receptors with diverse class I HD point mutations, the most common type of mutation found in T-ALL. 134
Conclusions and future directions Aberrant activation of the NOTCH signaling pathway plays a central role in T-ALL, a tumor in which multiple oncogenic and tumor suppressor pathways coordinately disrupt the regulatory programs controlling cell proliferation, differentiation and survival in normal developing thymocytes. The introduction of anti-NOTCH therapies in clinical trials may result in urgently needed improvements in therapy, particularly for patients with primary refractory and relapsed disease. Detailed correlative studies analyzing the genetic background of the tumors treated as well as elucidation of the mechanisms that mediate response to therapy and the genetic events implicated in resistance and relapse will be instrumental in defining the next steps towards achieving novel, more effective targeted treatments in this disease. Conflict of interest statement The Ferrando lab is partially funded by sponsored research projects funded by Merck and Pfizer on the pre-clinical testing of anti-NOTCH therapies in T-ALL. References [1] C.H. Pui, M.V. Relling, J.R. Downing. Acute lymphoblastic leukemia. N Engl J Med. 2004;350(15):1535-1548 Crossref. [2] A.J. Barrett, M.M. Horowitz, B.H. Pollock, M.J. Zhang, M.M. Bortin, G.R. Buchanan, et al. Bone marrow transplants from HLA-identical siblings as compared with chemotherapy for children with acute lymphoblastic leukemia in a second remission. N Engl J Med. 1994;331(19):1253-1258 Crossref. [3] J.C. Biggs, M.M. Horowitz, R.P. Gale, R.C. Ash, K. Atkinson, W. Helbig, et al. Bone marrow transplants may cure patients with acute leukemia never achieving remission with chemotherapy. Blood. 1992;80(4):1090-1093 [4] R. Dopfer, G. Henze, C. Bender-Gotze, W. Ebell, G. Ehninger, W. Friedrich, et al. Allogeneic bone marrow transplantation for childhood acute lymphoblastic leukemia in second remission after intensive primary and relapse therapy according to the BFM- and CoALL-protocols: results of the German Cooperative Study. Blood. 1991;78(10):2780-2784 [5] S.J. Forman, G.M. Schmidt, A.P. Nademanee, M.D. Amylon, N.J. Chao, J.L. Fahey, et al. Allogeneic bone marrow transplantation as therapy for primary induction failure for patients with acute leukemia. J Clin Oncol. 1991;9(9):1570-1574 [6] H. Schroeder, G. Gustafsson, U.M. Saarinen-Pihkala, A. Glomstein, G. Jonmundsson, K. Nysom, et al. Allogeneic bone marrow transplantation in second remission of childhood acute lymphoblastic leukemia: a population-based case control study from the Nordic countries. Bone Marrow Transplant. 1999;23(6):555-560 [7] D.I. Marks, E.M. Paietta, A.V. Moorman, S.M. Richards, G. Buck, G. DeWald, et al. T-cell acute lymphoblastic leukemia in adults: clinical features, immunophenotype, cytogenetics, and outcome from the large randomized prospective trial (UKALL XII/ECOG 2993). Blood. 2009;114(25):5136-5145 Crossref. [8] C.H. Pui, D. Pei, J.T. Sandlund, R.C. Ribeiro, J.E. Rubnitz, S.C. Raimondi, et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia. 2009;24(2):371-382 [9] A.A. Ferrando, A.T. Look. Clinical implications of recurring chromosomal and associated molecular abnormalities in acute lymphoblastic leukemia. Semin Hematol. 2000;37(4):381-395 Crossref. [10] A.A. Ferrando, D.S. Neuberg, J. Staunton, M.L. Loh, C. Huard, S.C. Raimondi, et al. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002;1(1):75-87 Crossref. [11] J. Soulier, E. Clappier, J.M. Cayuela, A. Regnault, M. Garcia-Peydro, H. Dombret, et al. HOXA genes are included in genetic and biologic networks defining human acute T-cell leukemia (T-ALL). Blood. 2005;106(1):274-286 Crossref. [12] A.A. Ferrando, S.A. Armstrong, D.S. Neuberg, S.E. Sallan, L.B. Silverman, S.J. Korsmeyer, et al. Gene expression signatures in MLL-rearranged T-lineage and B-precursor acute leukemias: dominance of HOX dysregulation. Blood. 2003;102(1):262-268 Crossref. [13] J. Hebert, J.M. Cayuela, J. Berkeley, F. Sigaux. Candidate tumor-suppressor genes MTS1 (p16INK4A) and MTS2 (p15INK4B) display frequent homozygous deletions in primary cells from T- but not from B-cell lineage acute lymphoblastic leukemias. Blood. 1994;84(12):4038-4044 [14] A.P. Weng, A.A. Ferrando, W. Lee, J.P.t. Morris, L.B. Silverman, C. Sanchez-Irizarry, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269-271 Crossref. [15] J.C. Aster, W.S. Pear, S.C. Blacklow. Notch signaling in leukemia. Annu Rev Pathol. 2008;3:587-613 Crossref. [16] A.A. Ferrando. The role of NOTCH1 signaling in T-ALL. Hematol Am Soc Hematol Educ Program. 2009;:353-361 Crossref. [17] K. De Keersmaecker, C. Graux, M.D. Odero, N. Mentens, R. Somers, J. Maertens, et al. Fusion of EML1 to ABL1 in T-cell acute lymphoblastic leukemia with cryptic t(9;14)(q34;q32). Blood. 2005;105(12):4849-4852 Crossref. [18] L.R. Finger, J. Kagan, G. Christopher, J. Kurtzberg, M.S. Hershfield, P.C. Nowell, et al. Involvement of the TCL5 gene on human chromosome 1 in T-cell leukemia and melanoma. Proc Natl Acad Sci USA. 1989;86(13):5039-5043 Crossref. [19] C.G. Begley, P.D. Aplan, S.M. Denning, B.F. Haynes, T.A. Waldmann, I.R. Kirsch. The gene SCL is expressed during early hematopoiesis and encodes a differentiation-related DNA-binding motif. Proc Natl Acad Sci USA. 1989;86(24):10128-10132 Crossref. [20] Q. Chen, J.T. Cheng, L.H. Tasi, N. Schneider, G. Buchanan, A. Carroll, et al. The tal gene undergoes chromosome translocation in T cell leukemia and potentially encodes a helix–loop–helix protein. EMBO J. 1990;9(2):415-424 [21] O. Bernard, P. Guglielmi, P. Jonveaux, D. Cherif, S. Gisselbrecht, M. Mauchauffe, et al. Two distinct mechanisms for the SCL gene activation in the t(1;14) translocation of T-cell leukemias. Genes Chromosom Cancer. 1990;1(3):194-208 Crossref. [22] Y. Xia, L. Brown, C.Y. Yang, J.T. Tsan, M.J. Siciliano, R. Espinosa III, et al. TAL2, a helix–loop–helix gene activated by the (7;9)(q34;q32) translocation in human T-cell leukemia. Proc Natl Acad Sci USA. 1991;88(24):11416-11420 Crossref. [23] J.D. Mellentin, S.D. Smith, M.L. Cleary. lyl-1, a novel gene altered by chromosomal translocation in T cell leukemia, codes for a protein with a helix–loop–helix DNA binding motif. Cell. 1989;58(1):77-83 Crossref. [24] J. Wang, S.N. Jani-Sait, E.A. Escalon, A.J. Carroll, P.J. de Jong, I.R. Kirsch, et al. The t(14;21)(q11.2;q22) chromosomal translocation associated with T-cell acute lymphoblastic leukemia activates the BHLHB1 gene. Proc Natl Acad Sci USA. 2000;97(7):3497-3502 Crossref. [25] E.A. McGuire, R.D. Hockett, K.M. Pollock, M.F. Bartholdi, S.J. O'Brien, S.J. Korsmeyer. The t(11;14)(p15;q11) in a T-cell acute lymphoblastic leukemia cell line activates multiple transcripts, including Ttg-1, a gene encoding a potential zinc finger protein. Mol Cell Biol. 1989;9(5):2124-2132 [26] J.M. Greenberg, T. Boehm, M.V. Sofroniew, R.J. Keynes, S.C. Barton, M.L. Norris, et al. Segmental and developmental regulation of a presumptive T-cell oncogene in the central nervous system. Nature. 1990;344(6262):158-160 Crossref. [27] T. Boehm, L. Foroni, Y. Kaneko, M.F. Perutz, T.H. Rabbitts. The rhombotin family of cysteine-rich LIM-domain oncogenes: distinct members are involved in T-cell translocations to human chromosomes 11p15 and 11p13. Proc Natl Acad Sci USA. 1991;88(10):4367-4371 Crossref. [28] B. Royer-Pokora, U. Loos, W.D. Ludwig. TTG-2, a new gene encoding a cysteine-rich protein with the LIM motif, is overexpressed in acute T-cell leukaemia with the t(11;14)(p13;q11). Oncogene. 1991;6(10):1887-1893 [29] A.J. Warren, W.H. Colledge, M.B. Carlton, M.J. Evans, A.J. Smith, T.H. Rabbitts. The oncogenic cysteine-rich LIM domain protein rbtn2 is essential for erythroid development. Cell. 1994;78(1):45-57 Crossref. [30] I.D. Dube, S. Kamel-Reid, C.C. Yuan, M. Lu, X. Wu, G. Corpus, et al. A novel human homeobox gene lies at the chromosome 10 breakpoint in lymphoid neoplasias with chromosomal translocation t(10;14). Blood. 1991;78(11):2996-3003 [31] M. Hatano, C.W. Roberts, M. Minden, W.M. Crist, S.J. Korsmeyer. Deregulation of a homeobox gene, HOX11, by the t(10;14) in T cell leukemia. Science. 1991;253(5015):79-82 [32] M. Lu, Z.Y. Gong, W.F. Shen, A.D. Ho. The tcl-3 proto-oncogene altered by chromosomal translocation in T-cell leukemia codes for a homeobox protein. EMBO J. 1991;10(10):2905-2910 [33] M.A. Kennedy, R. Gonzalez-Sarmiento, U.R. Kees, F. Lampert, N. Dear, T. Boehm, et al. HOX11, a homeobox-containing T-cell oncogene on human chromosome 10q24. Proc Natl Acad Sci USA. 1991;88(20):8900-8904 Crossref. [34] T.E. Hansen-Hagge, M. Schafer, H. Kiyoi, S.W. Morris, J.A. Whitlock, P. Koch, et al. Disruption of the RanBP17/Hox11L2 region by recombination with the TCRdelta locus in acute lymphoblastic leukemias with t(5;14)(q34;q11). Leukemia. 2002;16(11):2205-2212 Crossref. [35] S. Nagel, M. Kaufmann, H.G. Drexler, R.A. MacLeod. The cardiac homeobox gene NKX2-5 is deregulated by juxtaposition with BCL11B in pediatric T-ALL cell lines via a novel t(5;14)(q35.1;q32.2). Cancer Res. 2003;63(17):5329-5334 [36] G.K. Przybylski, W.A. Dik, P. Grabarczyk, J. Wanzeck, P. Chudobska, K. Jankowski, et al. The effect of a novel recombination between the homeobox gene NKX2-5 and the TRD locus in T-cell acute lymphoblastic leukemia on activation of the NKX2-5 gene. Haematologica. 2006;91(3):317-321 [37] F. Speleman, B. Cauwelier, N. Dastugue, J. Cools, B. Verhasselt, B. Poppe, et al. A new recurrent inversion, inv(7)(p15q34), leads to transcriptional activation of HOXA10 and HOXA11 in a subset of T-cell acute lymphoblastic leukemias. Leukemia. 2005;19(3):358-366 Crossref. [38] E.A. Shima, M.M. Le Beau, T.W. McKeithan, J. Minowada, L.C. Showe, T.W. Mak, et al. Gene encoding the alpha chain of the T-cell receptor is moved immediately downstream of c-myc in a chromosomal 8;14 translocation in a cell line from a human T-cell leukemia. Proc Natl Acad Sci USA. 1986;83(10):3439-3443 Crossref. [39] J. Erikson, L. Finger, L. Sun, A. ar-Rushdi, K. Nishikura, J. Minowada, et al. Deregulation of c-myc by translocation of the alpha-locus of the T-cell receptor in T-cell leukemias. Science. 1986;232(4752):884-886 [40] M. Urashima, H. Iyori, K. Fujisawa, Y. Hoshi, J. Akatsuka, K. Maekawa. Establishment and characteristics of a T-cell acute lymphoblastic leukemia cell line, JK-T1, with a chromosomal translocation between 8q24 and 14q13. Cancer Genet Cytogenet. 1992;64(1):86-90 Crossref. [41] T. Inaba, S. Murakami, N. Oku, K. Itoh, Y. Ura, S. Nakanishi, et al. Translocation between chromosomes 8q24 and 14q11 in T-cell acute lymphoblastic leukemia. Cancer Genet Cytogenet. 1990;49(1):69-74 Crossref. [42] E.A. Shima-Rich, A.M. Harden, T.W. McKeithan, J.D. Rowley, M.O. Diaz. Molecular analysis of the t(8;14)(q24;q11) chromosomal breakpoint junctions in the T-cell leukemia line MOLT-16. Genes Chromosom Cancer. 1997;20(4):363-371 Crossref. [43] E. Clappier, W. Cuccuini, A. Kalota, A. Crinquette, J.M. Cayuela, W.A. Dik, et al. The C-MYB locus is involved in chromosomal translocation and genomic duplications in human T-cell acute leukemia (T-ALL), the translocation defining a new T-ALL subtype in very young children. Blood. 2007;110(4):1251-1261 Crossref. [44] L.W. Ellisen, J. Bird, D.C. West, A.L. Soreng, T.C. Reynolds, S.D. Smith, et al. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell. 1991;66(4):649-661 Crossref. [45] T. Palomero, K.C. Barnes, P.J. Real, J.L. Bender, M.L. Sulis, V.V. Murty, et al. CUTLL1, a novel human T-cell lymphoma cell line with t(7;9) rearrangement, aberrant NOTCH1 activation and high sensitivity to gamma-secretase inhibitors. Leukemia. 2006;20(7):1279-1287 Crossref. [46] P.D. Aplan, D.P. Lombardi, A.M. Ginsberg, J. Cossman, V.L. Bertness, I.R. Kirsch. Disruption of the human SCL locus by “illegitimate” V-(D)-J recombinase activity. Science. 1990;250(4986):1426-1429 [47] P. Van Vlierberghe, M. van Grotel, H.B. Beverloo, C. Lee, T. Helgason, J. Buijs-Gladdines, et al. The cryptic chromosomal deletion del(11)(p12p13) as a new activation mechanism of LMO2 in pediatric T-cell acute lymphoblastic leukemia. Blood. 2006;108(10):3520-3529 Crossref. [48] I. Lahortiga, K. De Keersmaecker, P. Van Vlierberghe, C. Graux, B. Cauwelier, F. Lambert, et al. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat Genet. 2007;39(5):593-595 Crossref. [49] J. O'Neil, J. Tchinda, A. Gutierrez, L. Moreau, R.S. Maser, K.K. Wong, et al. Alu elements mediate MYB gene tandem duplication in human T-ALL. J Exp Med. 2007;204(13):3059-3066 Crossref. [50] M.H. Dreyling, J.A. Martinez-Climent, M. Zheng, J. Mao, J.D. Rowley, S.K. Bohlander. The t(10;11)(p13;q14) in the U937 cell line results in the fusion of the AF10 gene and CALM, encoding a new member of the AP-3 clathrin assembly protein family. Proc Natl Acad Sci USA. 1996;93(10):4804-4809 Crossref. [51] K.M. Carlson, C. Vignon, S. Bohlander, J.A. Martinez-Climent, M.M. Le Beau, J.D. Rowley. Identification and molecular characterization of CALM/AF10fusion products in T cell acute lymphoblastic leukemia and acute myeloid leukemia. Leukemia. 2000;14(1):100-104 Crossref. [52] V. Asnafi, I. Radford-Weiss, N. Dastugue, C. Bayle, D. Leboeuf, C. Charrin, et al. CALM-AF10 is a common fusion transcript in T-ALL and is specific to the TCRgammadelta lineage. Blood. 2003;102(3):1000-1006 Crossref. [53] D.S. Chervinsky, S.N. Sait, N.J. Nowak, T.B. Shows, P.D. Aplan. Complex MLL rearrangement in a patient with T-cell acute lymphoblastic leukemia. Genes Chromosom Cancer. 1995;14(1):76-84 Crossref. [54] J.E. Rubnitz, F.G. Behm, A.M. Curcio-Brint, R.P. Pinheiro, A.J. Carroll, S.C. Raimondi, et al. Molecular analysis of t(11;19) breakpoints in childhood acute leukemias. Blood. 1996;87(11):4804-4808 [55] P. Van Vlierberghe, M. van Grotel, J. Tchinda, C. Lee, H.B. Beverloo, P.J. van der Spek, et al. The recurrent SET-NUP214 fusion as a new HOXA activation mechanism in pediatric T-cell acute lymphoblastic leukemia. Blood. 2008;111(9):4668-4680 Crossref. [56] D.J. Hussey, M. Nicola, S. Moore, G.B. Peters, A. Dobrovic. The (4;11)(q21;p15) translocation fuses the NUP98 and RAP1GDS1 genes and is recurrent in T-cell acute lymphocytic leukemia. Blood. 1999;94(6):2072-2079 [57] C. Mecucci, R. La Starza, M. Negrini, S. Sabbioni, B. Crescenzi, P. Leoni, et al. t(4;11)(q21;p15) translocation involving NUP98 and RAP1GDS1 genes: characterization of a new subset of T acute lymphoblastic leukaemia. Br J Haematol. 2000;109(4):788-793 Crossref. [58] B. Tycko, S.D. Smith, J. Sklar. Chromosomal translocations joining LCK and TCRB loci in human T cell leukemia. J Exp Med. 1991;174(4):867-873 Crossref. [59] E. Clappier, W. Cuccuini, J.M. Cayuela, D. Vecchione, A. Baruchel, H. Dombret, et al. Cyclin D2 dysregulation by chromosomal translocations to TCR loci in T-cell acute lymphoblastic leukemias. Leukemia. 2006;20(1):82-86 Crossref. [60] K. Karrman, A. Andersson, H. Bjorgvinsdottir, B. Strombeck, C. Lassen, T. Olofsson, et al. Deregulation of cyclin D2 by juxtaposition with T-cell receptor alpha/delta locus in t(12;14)(p13;q11)-positive childhood T-cell acute lymphoblastic leukemia. Eur J Haematol. 2006;77(1):27-34 Crossref. [61] E. Flex, V. Petrangeli, L. Stella, S. Chiaretti, T. Hornakova, L. Knoops, et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med. 2008;205(4):751-758 Crossref. [62] C. Graux, J. Cools, C. Melotte, H. Quentmeier, A. Ferrando, R. Levine, et al. Fusion of NUP214 to ABL1 on amplified episomes in T-cell acute lymphoblastic leukemia. Nat Genet. 2004;36(10):1084-1089 Crossref. [63] M. Bar-Eli, H. Ahuja, A. Foti, M.J. Cline. N-RAS mutations in T-cell acute lymphocytic leukaemia: analysis by direct sequencing detects a novel mutation. Br J Haematol. 1989;72(1):36-39 Crossref. [64] M. Kawamura, H. Ohnishi, S.X. Guo, X.M. Sheng, M. Minegishi, R. Hanada, et al. Alterations of the p53, p21, p16, p15 and RAS genes in childhood T-cell acute lymphoblastic leukemia. Leuk Res. 1999;23(2):115-126 Crossref. [65] B.V. Balgobind, P. Van Vlierberghe, A.M. van den Ouweland, H.B. Beverloo, J.N. Terlouw-Kromosoeto, E.R. van Wering, et al. Leukemia-associated NF1 inactivation in patients with pediatric T-ALL and AML lacking evidence for neurofibromatosis. Blood. 2008;111(8):4322-4328 Crossref. [66] T. Palomero, M.L. Sulis, M. Cortina, P.J. Real, K. Barnes, M. Ciofani, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007;13(10):1203-1210 Crossref. [67] V. Tosello, M.R. Mansour, K. Barnes, M. Paganin, M.L. Sulis, S. Jenkinson, et al. WT1 mutations in T-ALL. Blood. 2009;114(5):1038-1045 Crossref. [68] M. Kleppe, I. Lahortiga, T. El Chaar, K. De Keersmaecker, N. Mentens, C. Graux, et al. Deletion of the protein tyrosine phosphatase gene PTPN2 in T-cell acute lymphoblastic leukemia. Nat Genet. 2010;42(6):530-535 Crossref. [69] P. Van Vlierberghe, T. Palomero, H. Khiabanian, J. Van der Meulen, M. Castillo, N. Van Roy, et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet. 2010;42(4):338-342 [70] C. Brou, F. Logeat, N. Gupta, C. Bessia, O. LeBail, J.R. Doedens, et al. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell. 2000;5(2):207-216 Crossref. [71] J.S. Mumm, R. Kopan. Notch signaling: from the outside in. Dev Biol. 2000;228(2):151-165 Crossref. [72] J.S. Mumm, E.H. Schroeter, M.T. Saxena, A. Griesemer, X. Tian, D.J. Pan, et al. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell. 2000;5(2):197-206 Crossref. [73] G. van Tetering, P. van Diest, I. Verlaan, E. van der Wall, R. Kopan, M. Vooijs. Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J Biol Chem. 2009;284(45):31018-31027 Crossref. [74] E.C. Bozkulak, G. Weinmaster. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol Cell Biol. 2009;29(21):5679-5695 Crossref. [75] K. Tanigaki, T. Honjo. Regulation of lymphocyte development by Notch signaling. Nat Immunol. 2007;8(5):451-456 Crossref. [76] K. Hozumi, C. Mailhos, N. Negishi, K. Hirano, T. Yahata, K. Ando, et al. Delta-like 4 is indispensable in thymic environment specific for T cell development. J Exp Med. 2008;205(11):2507-2513 Crossref. [77] H. Han, K. Tanigaki, N. Yamamoto, K. Kuroda, M. Yoshimoto, T. Nakahata, et al. Inducible gene knockout of transcription factor recombination signal binding protein-J reveals its essential role in T versus B lineage decision. Int Immunol. 2002;14(6):637-645 [78] F. Radtke, A. Wilson, G. Stark, M. Bauer, J. van Meerwijk, H.R. MacDonald, et al. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity. 1999;10(5):547-558 Crossref. [79] J.C. Pui, D. Allman, L. Xu, S. DeRocco, F.G. Karnell, S. Bakkour, et al. Notch1 expression in early lymphopoiesis influences B versus T lineage determination. Immunity. 1999;11(3):299-308 Crossref. [80] M. Ciofani, J.C. Zuniga-Pflucker. A survival guide to early T cell development. Immunol Res. 2006;34(2):117-132 Crossref. [81] A. Wolfer, A. Wilson, M. Nemir, H.R. MacDonald, F. Radtke. Inactivation of Notch1 impairs VDJbeta rearrangement and allows pre-TCR-independent survival of early alpha beta Lineage Thymocytes. Immunity. 2002;16(6):869-879 Crossref. [82] B. Reizis, P. Leder. Direct induction of T lymphocyte-specific gene expression by the mammalian Notch signaling pathway. Genes Dev. 2002;16(3):295-300 Crossref. [83] S. Gonzalez-Garcia, M. Garcia-Peydro, E. Martin-Gayo, E. Ballestar, M. Esteller, R. Bornstein, et al. CSL-MAML-dependent Notch1 signaling controls T lineage-specific IL-7R{alpha} gene expression in early human thymopoiesis and leukemia. J Exp Med. 2009;206(4):779-791 Crossref. [84] A.P. Weng, J.M. Millholland, Y. Yashiro-Ohtani, M.L. Arcangeli, A. Lau, C. Wai, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006;20(15):2096-2109 Crossref. [85] M. Ciofani, J.C. Zuniga-Pflucker. Notch promotes survival of pre-T cells at the beta-selection checkpoint by regulating cellular metabolism. Nat Immunol. 2005;6(9):881-888 Crossref. [86] W.S. Pear, J.C. Aster, M.L. Scott, R.P. Hasserjian, B. Soffer, J. Sklar, et al. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med. 1996;183(5):2283-2291 Crossref. [87] L. Girard, Z. Hanna, N. Beaulieu, C.D. Hoemann, C. Simard, C.A. Kozak, et al. Frequent provirus insertional mutagenesis of Notch1 in thymomas of MMTVD/myc transgenic mice suggests a collaboration of c-myc and Notch1 for oncogenesis. Genes Dev. 1996;10(15):1930-1944 Crossref. [88] M.J. Malecki, C. Sanchez-Irizarry, J.L. Mitchell, G. Histen, M.L. Xu, J.C. Aster, et al. Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Mol Cell Biol. 2006;26(12):4642-4651 Crossref. [89] M.L. Sulis, O. Williams, T. Palomero, V. Tosello, S. Pallikuppam, P.J. Real, et al. NOTCH1 extracellular juxtamembrane expansion mutations in T-ALL. Blood. 2008;112(3):733-740 Crossref. [90] W.R. Gordon, M. Roy, D. Vardar-Ulu, M. Garfinkel, M.R. Mansour, J.C. Aster, et al. Structure of the Notch1-negative regulatory region: implications for normal activation and pathogenic signaling in T-ALL. Blood. 2009;113(18):4381-4390 Crossref. [91] B.J. Thompson, S. Buonamici, M.L. Sulis, T. Palomero, T. Vilimas, G. Basso, et al. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med. 2007;204(8):1825-1835 Crossref. [92] J. O'Neil, J. Grim, P. Strack, S. Rao, D. Tibbitts, C. Winter, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007;204(8):1813-1824 Crossref. [93] S. Akhoondi, D. Sun, N. von der Lehr, S. Apostolidou, K. Klotz, A. Maljukova, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67(19):9006-9012 Crossref. [94] A.C. Minella, B.E. Clurman. Mechanisms of tumor suppression by the SCF(Fbw7). Cell Cycle. 2005;4(10):1356-1359 Crossref. [95] Y. Fujii, M. Yada, M. Nishiyama, T. Kamura, H. Takahashi, R. Tsunematsu, et al. Fbxw7 contributes to tumor suppression by targeting multiple proteins for ubiquitin-dependent degradation. Cancer Sci. 2006;97(8):729-736 Crossref. [96] J.H. Mao, I.J. Kim, D. Wu, J. Climent, H.C. Kang, R. DelRosario, et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008;321(5895):1499-1502 Crossref. [97] M.Y. Chiang, L. Xu, O. Shestova, G. Histen, S. L'Heureux, C. Romany, et al. Leukemia-associated NOTCH1 alleles are weak tumor initiators but accelerate K-ras-initiated leukemia. J Clin Invest. 2008;118(9):3181-3194 Crossref. [98] T. Palomero, W.K. Lim, D.T. Odom, M.L. Sulis, P.J. Real, A. Margolin, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci USA. 2006;103(48):18261-18266 Crossref. [99] A.A. Margolin, T. Palomero, P. Sumazin, A. Califano, A.A. Ferrando, G. Stolovitzky. ChIP-on-chip significance analysis reveals large-scale binding and regulation by human transcription factor oncogenes. Proc Natl Acad Sci USA. 2009;106(1):244-249 Crossref. [100] V.M. Sharma, J.A. Calvo, K.M. Draheim, L.A. Cunningham, N. Hermance, L. Beverly, et al. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol Cell Biol. 2006;26(21):8022-8031 Crossref. [101] S.M. Chan, A.P. Weng, R. Tibshirani, J.C. Aster, P.J. Utz. Notch signals positively regulate activity of the mTOR pathway in T-cell acute lymphoblastic leukemia. Blood. 2007;110(1):278-286 Crossref. [102] H. Sade, S. Krishna, A. Sarin. The anti-apoptotic effect of Notch-1 requires p56lck-dependent, Akt/PKB-mediated signaling in T cells. J Biol Chem. 2004;279(4):2937-2944 [103] I. Joshi, L.M. Minter, J. Telfer, R.M. Demarest, A.J. Capobianco, J.C. Aster, et al. Notch signaling mediates G1/S cell-cycle progression in T cells via cyclin D3 and its dependent kinases. Blood. 2009;113(8):1689-1698 Crossref. [104] T. Dohda, A. Maljukova, L. Liu, M. Heyman, D. Grander, D. Brodin, et al. Notch signaling induces SKP2 expression and promotes reduction of p27Kip1 in T-cell acute lymphoblastic leukemia cell lines. Exp Cell Res. 2007;313(14):3141-3152 Crossref. [105] S.S. Rao, J. O'Neil, C.D. Liberator, J.S. Hardwick, X. Dai, T. Zhang, et al. Inhibition of NOTCH signaling by gamma secretase inhibitor engages the RB pathway and elicits cell cycle exit in T-cell acute lymphoblastic leukemia cells. Cancer Res. 2009;69(7):3060-3068 Crossref. [106] E. Sicinska, I. Aifantis, L. Le Cam, W. Swat, C. Borowski, Q. Yu, et al. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell. 2003;4(6):451-461 Crossref. [107] L.L. Song, Y. Peng, J. Yun, P. Rizzo, V. Chaturvedi, S. Weijzen, et al. Notch-1 associates with IKKalpha and regulates IKK activity in cervical cancer cells. Oncogene. 2008;27(44):5833-5844 Crossref. [108] H.M. Shin, L.M. Minter, O.H. Cho, S. Gottipati, A.H. Fauq, T.E. Golde, et al. Notch1 augments NF-kappaB activity by facilitating its nuclear retention. EMBO J. 2006;25(1):129-138 Crossref. [109] T. Vilimas, J. Mascarenhas, T. Palomero, M. Mandal, S. Buonamici, F. Meng, et al. Targeting the NF-kappaB signaling pathway in Notch1-induced T-cell leukemia. Nat Med. 2007;13(1):70-77 Crossref. [110] S. Breit, M. Stanulla, T. Flohr, M. Schrappe, W.D. Ludwig, G. Tolle, et al. Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood. 2006;108(4):1151-1157 Crossref. [111] M.J. Park, T. Taki, M. Oda, T. Watanabe, K. Yumura-Yagi, R. Kobayashi, et al. FBXW7 and NOTCH1 mutations in childhood T cell acute lymphoblastic leukaemia and T cell non-Hodgkin lymphoma. Br J Haematol. 2009;145(2):198-206 Crossref. [112] V. Asnafi, A. Buzyn, S. Le Noir, F. Baleydier, A. Simon, K. Beldjord, et al. NOTCH1/FBXW7 mutation identifies a large subgroup with favorable outcome in adult T-cell acute lymphoblastic leukemia (T-ALL): a Group for Research on Adult Acute Lymphoblastic Leukemia (GRAALL) study. Blood. 2009;113(17):3918-3924 Crossref. [113] M. van Grotel, J.P. Meijerink, H.B. Beverloo, A.W. Langerak, J.G. Buys-Gladdines, P. Schneider, et al. The outcome of molecular-cytogenetic subgroups in pediatric T-cell acute lymphoblastic leukemia: a retrospective study of patients treated according to DCOG or COALL protocols. Haematologica. 2006;91(9):1212-1221 [114] M.R. Mansour, M.L. Sulis, V. Duke, L. Foroni, S. Jenkinson, K. Koo, et al. Prognostic implications of NOTCH1 and FBXW7 mutations in adults with T-cell acute lymphoblastic leukemia treated on the MRC UKALLXII/ECOG E2993 protocol. J Clin Oncol. 2009;27(26):4352-4356 Crossref. [115] L. Zuurbier, V. Homminga IC, M.L. te Winkel, J. Buijs-Gladdines, C. Kooi, W.K. Smits, et al. NOTCH1 and/or FBXW7 mutations predict for initial good prednisone response but not for improved outcome in pediatric T-cell Acute Lymphoblastic Leukemia patients treated on DCOG or COALL protocols. Leukemia. 2010;24(12):2014-2022 Crossref. [116] E. Clappier, S. Collette, N. Grardel, S. Girard, L. Suarez, G. Brunie, et al. NOTCH1 and FBXW7 mutations have a favorable impact on early response to treatment but not on outcome in children with T-cell acute lymphoblastic (T-ALL) leukemia treated on EORTC trials 58881 and 58951. Leukemia. 2010;24(12):2323-2331 [117] K.Z. Kox, M. Stanulla, S. Leibe, M. Schrappe, W.D. Ludwig, R. Koehler, et al. The favorable effect of activating NOTCH1 receptor mutations on long-term outcome in T-ALL patients treated on the ALL-BFM 2000 protocol can be separated from FBXW7 loss of function. Leukemia. 2010;24(12):2003-2004 [118] G. Evin, M.F. Sernee, C.L. Masters. Inhibition of gamma-secretase as a therapeutic intervention for Alzheimer's disease: prospects, limitations and strategies. CNS Drugs. 2006;20(5):351-372 Crossref. [119] D. Selkoe, R. Kopan. Notch and Presenilin: regulated intramembrane proteolysis links development and degeneration. Annu Rev Neurosci. 2003;26:565-597 Crossref. [120] D. Deangelo, R. Stone, L. Silverman, W. Stock, E. Attar, I. Fearen, et al. A phase I clinical trial of the notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. in: Journal of Clinical Oncology, 2006 ASCO Annual Meeting Proceedings Part I. 24(18S) (, 2006) 6585 [121] J.H. van Es, M.E. van Gijn, O. Riccio, M. van den Born, M. Vooijs, H. Begthel, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435(7044):959-963 Crossref. [122] O. Riccio, M.E. van Gijn, A.C. Bezdek, L. Pellegrinet, J.H. van Es, U. Zimber-Strobl, et al. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27(Kip1) and p57(Kip2). EMBO Rep. 2008;9(4):377-383 Crossref. [123] J. Milano, J. McKay, C. Dagenais, L. Foster-Brown, F. Pognan, R. Gadient, et al. Modulation of notch processing by gamma-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci. 2004;82(1):341-358 Crossref. [124] J. Tammam, C. Ware, C. Efferson, J. O'Neil, S. Rao, X. Qu, et al. Down-regulation of the Notch pathway mediated by a gamma-secretase inhibitor induces anti-tumour effects in mouse models of T-cell leukaemia. Br J Pharmacol. 2009;158(5):1183-1195 Crossref. [125] Cerchietti LC, Ghetu AF, Zhu X, Da Silva GF, Zhong S, Matthews M, et al. A small-molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer Cell. 17(4):400–411. [126] K. Cullion, K.M. Draheim, N. Hermance, J. Tammam, V.M. Sharma, C. Ware, et al. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood. 2009;113(24):6172-6181 Crossref. [127] K. De Keersmaecker, I. Lahortiga, N. Mentens, C. Folens, L. Van Neste, S. Bekaert, et al. In vitro validation of gamma-secretase inhibitors alone or in combination with other anti-cancer drugs for the treatment of T-cell acute lymphoblastic leukemia. Haematologica. 2008;93(4):533-542 Crossref. [128] P.J. Real, V. Tosello, T. Palomero, M. Castillo, E. Hernando, E. de Stanchina, et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med. 2009;15(1):50-58 Crossref. [129] P.J. Real, A.A. Ferrando. NOTCH inhibition and glucocorticoid therapy in T-cell acute lymphoblastic leukemia. Leukemia. 2009;23(8):1374-1377 Crossref. [130] Wei P, Walls M, Qiu M, Ding R, Denlinger RH, Wong A, et al. Evaluation of selective gamma-secretase inhibitor PF-03084014 for its antitumor efficacy and gastrointestinal safety to guide optimal clinical trial design. Mol Cancer Ther. 9(6):1618–1628. [131] H. Medyouf, X. Gao, F. Armstrong, S. Gusscott, Q. Liu, A.L. Gedman, et al. Acute T-cell leukemias remain dependent on Notch signaling despite PTEN and INK4A/ARF loss. Blood. 2010;115(6):1175-1184 Crossref. [132] R.E. Moellering, M. Cornejo, T.N. Davis, C. Del Bianco, J.C. Aster, S.C. Blacklow, et al. Direct inhibition of the NOTCH transcription factor complex. Nature. 2009;462(7270):182-188 Crossref. [133] Y. Wu, C. Cain-Hom, L. Choy, T.J. Hagenbeek, G.P. de Leon, Y. Chen, et al. Therapeutic antibody targeting of individual Notch receptors. Nature. 2010;464(7291):1052-1057 Crossref. [134] M. Aste-Amezaga, N. Zhang, J.E. Lineberger, B.A. Arnold, T.J. Toner, et al. Characterization of Notch1 antibodies that inhibit signaling of both normal and mutated Notch1 receptors. PLoS One. 2010;5(2):e9094 Crossref. Footnotes a Institute for Cancer Genetics, Columbia University, NY, USA b Department of Pediatrics, Columbia University Medical Center, NY, USA c Department of Pathology, Columbia University Medical Center, NY, USA lowast Corresponding
0 notes
Text
Frequency and geographic distribution of TERT promoter mutations in primary hepatocellular carcinoma
Abstract
Primary hepatocellular carcinoma (HCC) mainly develops in subjects chronically infected with hepatitis B (HBV) and C (HCV) viruses through a multistep process characterized by the accumulation of genetic alterations in the human genome. Nucleotide changes in coding regions (i.e. TP53, CTNNB1, ARID1A and ARID2) as well as in non-coding regions (i.e. TERT promoter) are considered cancer drivers for HCC development with variable frequencies in different geographic regions depending on the etiology and environmental factors. Recurrent hot spot mutations in TERT promoter (G > A at-124 bp; G > A at −146 bp), have shown to be common events in many tumor types including HCC and to up regulate the expression of telomerases. We performed a comprehensive review of the literature evaluating the differential distribution of TERT promoter mutations in 1939 primary HCC from four continents. Mutation rates were found higher in Europe (56.6%) and Africa (53.3%) than America (40%) and Asia (42.5%). In addition, HCV-related HCC were more frequently mutated (44.8% in US and 69.7% in Asia) than HBV-related HCC (21.4% in US and 45.5% in Africa). HCC cases associated to factors other than hepatitis viruses are also frequently mutated in TERT promoter (43.6%, 52.6% and 57.7% in USA, Asia and Europe, respectively). These results support a major role for telomere elongation in HCV-related and non-viral related hepatic carcinogenesis and suggest that TERT promoter mutations could represent a candidate biomarker for the early detection of liver cancer in subjects with HCV infection or with metabolic liver diseases.
http://ift.tt/2qAmJYf
0 notes
Text
Somatic mutations in murine models of leukemia and lymphoma: Disease specificity and clinical relevance
Abstract
Malignant transformation is a multistep process that is dictated by acquisition of multiple genomic aberrations that provide growth and survival advantage. During the post genomic era, high throughput genomic sequencing has advanced exponentially, leading to identification of countless cancer associated mutations with potential for targeted therapy. Mouse models of cancer serve as excellent tools to examine the functionality of gene mutations and their contribution to the malignant process. However, it remains unclear whether the genetic events that occur during transformation are similar in mice and humans. To address that, we chose several transgenic mouse models of hematopoietic malignancies and identified acquired mutations in these mice by means of targeted re-sequencing of known cancer-associated genes as well as whole exome sequencing. We found that mutations that are typically found in acute myeloid leukemia or T cell acute lymphoblastic leukemia patients are also common in mouse models of the respective disease. Moreover, we found that the most frequent mutations found in a mouse model of lymphoma occur in a set of epigenetic modifier genes, implicating this pathway in the generation of lymphoma. These results demonstrate that genetically engineered mouse models (GEMM) mimic the genetic evolution of human cancer and serve as excellent platforms for target discovery and validation. This article is protected by copyright. All rights reserved.
http://ift.tt/2lNpyRe
0 notes