
This is a blog dedicated to spreading awareness for those with invisible diseases and disablities, specifically Cystic Fibrosis.
Don't wanna be here? Send us removal request.
Statistics
We looked inside some of the posts by learningtobreathe2019 and here's what we found interesting.
Average Info
Notes Per Post
1
Likes Per Post
1
Reblog Per Post
0
Reply Per Post
0
Time Between Posts
9 days
Number of Posts By Type
Text
7
Photo
2
Last Seen Tumblr Blogs





Fun Fact
Tumblr has 16.74 million mobile monthly users in the US.
Text
0 notes
Text

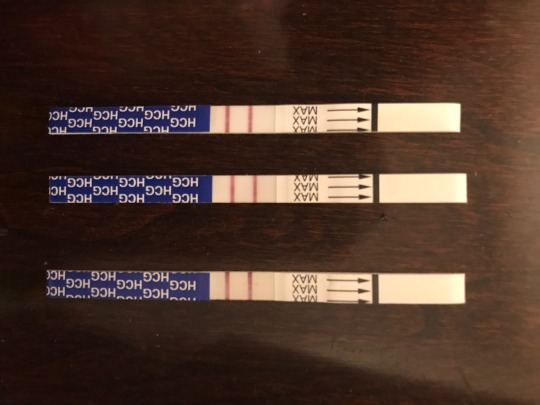

Post 6 Pregnancy and the possibilities of CF
4/24/2020
(Pictures are of my wife and son the day she found out she was pregnant again)
Within the last week, my wife and I have found out that we are being blessed again with another wonderful baby as my wife is pregnant.
I wanted to thus take this opportunity to write a little bit on Cystic Fibrosis and how parents who are both recessive carriers (like my wife and I) will always have a chance to produce a baby with CF.
My wife and I are both recessive carriers of the Cystic Fibrosis gene. Thus with the genetic possibilities when we create a child, each of our children will always have a 25% chance of actively having the disease like my son William. Also they will have a 50% chance of being a recessive carrier, and a 25% chance of not having or carrying the disease at all.
This was one of the hardest issues to discuss when we first found out about Williams diagnosis- should we continue to have more children.
After a short conversation we easily decided we would. With a 75% chance of not being actively affected by the disease any future children we have are more likely than not, to not be affected. With this in mind and with our personal spiritual beliefs we both decided that God would provide and protect us with whatever may come, and strengthen us for whatever that is in our future.
Thankfully as of now, it is another child.
Throughout this pregnancy we will be praying for the health of this baby and we will love it no more nor less regardless of the health issues it may or may not have upon birth.
0 notes
Text
Post 5, April 15th 2020
As of today there are still states that have written mandates stating they can withhold lifesaving care to patients with CF- Louisiana and Utah. These mandates are extremely outdated and were written when CF knowledge was in its infancy and was considered a death sentence. CF research and treatments have grown exponentially since it’s discovery, yet some states still have these antiquated and ridiculous laws that would prevent my child from getting the care needed to save his life, simply because he has a disease.
Other states have differently worded mandates allowing them to withhold ventilators if a shortage occurs during a crisis like Covid 19- Oklahoma, Wisconsin, Florida, Vermont, and Alaska.
In a capitalist country with a single payer funded health care system, ie im paying for my health care needs not the government, it is wrong for them to withhold services from my son simply because he has an autoimmune disease.
Praying for these laws and rules to be overturned for those suffering in those states with CF. And also praying for everyone’s safety during this time.
Michael
0 notes
Text
Post 4 Dealing With Covid-19
4/10/2020
On March 16th my family and I moved from Washington State to Missouri. This move was for a transfer in jobs within the Army. While traveling during this outbreak we had to be extra cautious for William. When we left Washington we filled up with Gas on base and did not stop again until we reached a very remote gas station in Oregon and before I got back in the car I practically bathed in hand sanitizer. This was repeated the entire way to Missouri and we had packed and prepped food prior to leaving so that we did not have to go into the gas stations for food.
Each night when we arrived at our hotel my wife would go in first while I stayed in the car with William. My wife would completely disinfect everything as best as possible before we brought William in. When we finally arrived in Missouri she did the same with our apartment.
I was made to go into quarantine when I checked into work in Missouri because I came from Washington State. Now that my quarantine is over I am deemed “essential” and have had to report to work everyday on base.
Immediately upon returning home from work I completely undress as soon as I enter my apartment and put all of my uniform in a garbage bag. I then go shower and attempt to disinfect myself as best as possible. I then go and wash my uniforms every night after work.
We feel safer here in Missouri than we did in Washington because of how close we were to the west coast epicenter there.
Many of these social distancing steps that the world has implemented now and sanitary guidelines have been adopted by Cystic Fibrosis families for years. It is a lifestyle that we have to live with forever and that is okay because it is what we have to do to keep our loved ones safe.
I hope everyone is remaining safe,
Michael.
0 notes
Text
Post 3
3/30/2020
This post is going to cover my wife’s perspective in regards to our son’s diagnosis with Cystic Fibrosis. I started to try and write this by myself from what I knew her experiences were, but I found that was not as easy as I thought and did not seem as interesting to read. Thus I decided to use the question and answer format for this post. I asked the questions and the responses and answers are my wife’s. I hope you enjoy the read.
Q1: Did you have any idea during your pregnancy or after William was born that he may have a disability?
A1: No. I had done a few standard blood tests that most pregnant women get that rule out things like cystic fibrosis. I was negative for the most common CF gene mutation as well as signs for other issues. When he was born, his vitals were great and he looked perfectly healthy so I had no idea.
Q2: What were your thoughts and feelings during William’s repeated blood tests in the hospital in the days after he was born? Were you worried or did you just think it was an error during testing?
A2: I was worried very little at the beginning because I had no reason to think he may have CF. As far as we both know, no one on either side of the family had CF. As each blood test came back slightly elevated, I started to worry a little more. The doctor though reassured us that these blood tests are very basic and can be inaccurate. He said that these blood tests on newborns can come back elevated but often level out after about a week. So, more in-depth testing would have to be done to actually determine if he had cystic fibrosis or not.
Q3: After the blood tests continually came back elevated and William had to begin getting sweat tests done, did you begin to worry?
A3: Like I mentioned a minute ago I started to worry more and more with each blood test. I wasn’t panicking though. Deep down I felt like he didn’t have CF and the sweat test would prove that.
Q4: After the first inconclusive sweat test how did you feel?
A4: I was very frustrated and honestly, pretty pissed. I was supposed to be at home loving on my new baby and healing from a csection. Instead, I was sent to an understaffed special lab and forced to sit in a heated room with my newborn baby bundled up in a fleece hat and multiple blankets. The thought of having to put him through another sweat test was heartbreaking, especially because I didn’t think he had cystic fibrosis.
Q5: On the day we first found out Willie had CF, when I handed the phone to you, what were Dr. Friedman’s exact words if you can remember?
A5: He said, “William’s sweat test came back positive. So, he does have cystic fibrosis. The results for the test are from 1 to 100 and anything over 50 is considered positive for cystic fibrosis. William came in at 68.” I’m pretty sure he was still talking after that but I don’t remember. I heard that he had cystic fibrosis. I kind of went numb after that and I remember just putting the phone down pulling Willie to my chestvand just crying.
Q6: What were you feeling when you first realized and comprehended that Willie had CF?
A6: I was scared and heartbroken. At the time, I thought CF was a death sentence and he wouldn’t live past his teens or early 20s. I didn’t want him to live hooked up to oxygen or grow up waiting for a lung transplant.
Q7: What was the hardest part in that first month after his diagnosis?
A7: Two things. Because we didn’t know his pancreas worked yet or not, I had to give him enzymes every time I breastfed. I was horrible at remembering to give him the pellets in the middle of the night. The other thing that was hard was full grasping that this didn’t mean he would die young.
Q8:What has been the hardest part since that first month?
A8: Hands down his breathing treatments. He hates them and cries and screams when he has to do them. I do one every morning and one every night. The morning one is hyper saline and takes about 45 mins and the one at night only takes about 15 so it’s not as bad. It’s so sad because I know this what I have to do to make sure he can live a long healthy life but I hate making him cry like he does.
Q9: How has having a son with CF affected how you view life?
A9: Wow, thats a loaded question so I’ll just give you the jist. I’m more grateful for the health I have personally. It has caused me to think a lot more about death and our vulnerability as humans. We aren’t promised tomorrow or an easy life at that.
Q10: How has having a son with CF affected your outlook on the Covid-19 situation? Has it affected your daily life differently than if he had not had CF?
A10: Oh my gosh has it affected my outlook! I’m concerned about you and I getting it but I feel like we’d be fine. William on the other hand is considered a high risk patient because of his CF. I think I’d be worried for him even without him having CF but because he does have CF, I’m hyper aware of germs anyway and this just takes it to a new level. It makes me OCD and anxious about every little detail. I know my daily life right now with a CF son and COVID-19, I go above and beyond most. I do my best to sanitize everything but also carry around hand sanitizer and disinfectant wipes just in case I need them.
0 notes
Text
Post 2- Learning about Cystic Fibrosis and Dealing with Anger-2/22/2020
The five stages that a person commonly experiences when grieving the loss of another or when dealing with bad news are denial, then anger, followed by bargaining, depression and finally acceptance. Once my wife and I got off the phone with Dr. Friedman, our son’s pulmonologist, and eventually stopped crying, we progressed into the anger phase of grieving rather quickly. Prior to receiving the news; however, we had been in denial. Honestly, we each agreed to avoid doing any research on Cystic Fibrosis despite the warning signs our son was showing during the testing phase. We had told ourselves, for the three short weeks that he had been alive, “He can’t have it”, “God wouldn’t do that to an innocent child”, “He looks too healthy”, “No, not our son”, and we had refused to believe it was even possible. Thus, we practically convinced ourselves and each other there was zero possibility our son had CF.
Individually and without the other knowing, my wife and I had both given into temptation and took a quick spin around the dance floor with Google. Little did we know, neither of us would make it past the very first search page. My wife and I had both exited the initial Google page after seeing the very first article’s synopsis, which stated that the average life expectancy of an American with Cystic Fibrosis was just 37 years. Just 37 years! Why would we roll the dice again and risk falling into the rabbit hole that is Google. Thus, when we did inevitably receive the news about our little boy, all we could focus on was that dreaded number. The number 37 was burned into our minds and hearts because at that very moment all it meant was early mortality for our newborn son and pain that we would experience when losing a child. We both refused to do anymore research on CF that day. We were mad at life, angry with God and just simply confused as to what we must have done wrong. How could such an innocent and perfect little human deserve such a curse? Despite our flood of emotions, my wife and I chose to dive head first into the deep abyss of online research. Through our research, we learned that “Cystic Fibrosis is a hereditary disease that affects the lungs and digestive system. The body produces thick and sticky mucus that can clog the lungs and obstruct the pancreas” (McIntosh). In the days that followed that dreaded call, we spent countless hours in a sterile clinic room with the pulmonologists at the Children’s hospital. We asked as many questions as we could possibly think of. The two of us would absorb and comprehend as much of the information being thrown at us, as humanly possible. We were still angry but we needed to know about the disease. What was going on with our son’s body? What would this mean for his future? After nearly a dozen CF specific appointments with doctors in several different specialties we learned a lot, shed more tears, shared embraces with doctors we had just met and loved on our baby boy with hope of a bright future. As a couple, we continued to read everything about Cystic Fibrosis that we could get our hands on. We took our fair share of deep breaths as we cried, we dealt with physical chest pain and stomach aches caused by the abundance of emotional pain. Our hearts were broken for this perfect little being that we loved so deeply.
During those days and weeks of repetitive hospital visits and long nights in front of our computer screens, we would learn and grow exponentially as a family. So what did we learn? Well, we found out that my wife and I are both carriers of a recessive CF gene. This means that neither of us actually have CF but that every one of our children will be born with a 25% chance of having Cystic Fibrosis. We also learned that Cystic Fibrosis is not just a one size fits all diagnosis. This is good and bad. Cystic Fibrosis has thousands of possible genetic mutations that cause the disease to affect its hosts in different manners. My son for instance, has one of the most common CF gene mutations, which he inherited from me, and his other mutation is one of the most rare, with only 8 other cases every documented in world. The rare gene was inherited from my wife. The downside of the common mutation I passed to our son is often the cause of a more severe form of CF. On the plus side, the extremely rare gene my wife passed to our son, causes the more severe gene to mellow out in a way. His rare gene mutation would bless us with a bit of hope. This gene caused him to be classified a pancreatic sufficient, unlike the majority of those with Cystic Fibrosis. This means that his body is capable of properly digesting the food he eats and thus turning it into nutrients. Unlike my son’s case, those who are pancreatic insufficient, have to eat a ridiculous amount of calories every day to meet their caloric needs. These people often have to intake double, sometimes even triple the amount of calories each day as well as taking medication to help with nutrient absorption. We also learned that his rare mixture of CF genes, thankfully, allows my son to qualify for what is called a modulator once he reaches the FDA approved age. From what we have been told, these medications or modulators are pills that help the body almost completely counteract the disease. The medication, when taken daily, will essentially counteract a person’s CF genes and allow them to live a more normal life. The specific modulator that my son qualifies for is called Trikafta. At the time of my son’s diagnosis, FDA approval was still being sought after for CF patients 12 years and older. As of the last few months, Trikafta was approved for those 12 and older and new trials for Cystic Fibrosis patients 6 to 11 have started. We are being told that trials for children with CF ages 2-5 will also begin within the next year. This is nothing short of a blessing to our family.
After one of the worst days of our lives we were now slightly more informed and hopeful. Even with that being said, we were still very much in the anger phase of grieving. The anger phase lasted for quite a while and although we feel like we are moving past the anger we still find ourselves holding onto it. My wife and I both find ourselves harboring anger towards the universe and life as a whole. We must consciously reign our emotions in and counter act that anger with positive hope and thoughts of somewhat normal future for our son.
I will post again next week but that post will be about my wife. I’ll cover things from her perspective and her emotional journey from diagnosis until now. I hope that you all have enjoyed reading about our emotional battles and have been able to learn something about an invisible disease called Cystic Fibrosis. As always, if you have any critiques or criticisms I would love to hear your feedback.
References
Cystic Fibrosis: Symptoms, Causes, and Management
James McIntosh - https://www.medicalnewstoday.com/articles/147960
0 notes
Photo



July 8th 2019. These photos were taken shortly after we found out William had Cystic Fibrosis and we had finally stopped crying for a few brief moments.
Final part of Post 1, 2/8/2020
0 notes
Photo


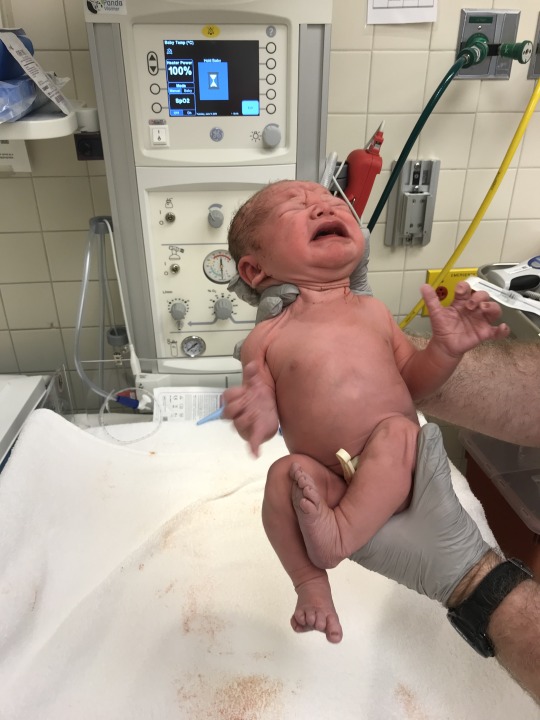


June 11th, 2019. The day William was born.
Part of Post 1. 2/8/2020
0 notes
Text
Post 1, Birth and Diagnosis, 2/8/2020
Greetings and welcome to the Learning to Breathe Blog, I am the creator and moderator, Michael Krystallis. This blog is being created to, and is dedicated to, spreading awareness for those with invisible diseases and disabilities, specifically to my infant son who was diagnosed with Cystic Fibrosis at just 3 weeks old. My plan is for this blog to be relevant and engaging to all who either have loved ones with “invisible diseases” or have no clue what the term even means. I wanted to create this blog so that it may serve as a resource to others whom may be affected emotionally, physically or spiritually by an invisible disease or disability. Personally I hope this blog reaches others and helps others see what life is like for small children diagnosed with Cystic Fibrosis specifically, but also any invisible illness. I am sure it will also serve as a therapeutic outlet for me as well and allow me to express and put into words what my family and my son have gone through and will continue to go through for the rest of our lives since June 11th 2019.
You meet the love of your life, you get married and you start a family. It’s that simple, right? No one ever mentions how hard the last part of that statement can be. My wife and I both knew, at very early ages, that we wanted to be parents. So, as soon as we were married we started trying for a family. We would have never guessed that those months of trying would quickly turn into years. We wanted to be parents so badly that during this time, we also decided to pursue adoption. First, we would have our hand at being matched with an expectant mother because we really wanted a newborn. After many cases being presented to us, we decided maybe we were being led to adopt from foster care. For various reasons, things were taking longer than we had hoped but fate was on our side. After almost two years of trying we decided something was wrong. One of us, both of us, something just wasn’t working like it was supposed to. So, with heavy hearts we called a prestigious local fertility clinic and scheduled our first appointment. The day before our appointment, my wife would give me the best birthday present to this day. The morning before our infertility appointment, my wife took her last pregnancy test and it was positive. Finally, after several years, after all the heart break month after month, we were going to be parents. We couldn’t have been more excited.
On Sunday, June 9th, just 3 days before her due date, my very pregnant wife curled up in bed with what she thought was a upset stomach from something she had eaten. I’m sure you can guess where this is going. Nope, she didn’t have an upset stomach, she was in labor. After making our way to the hospital, lots of ups and downs and thirty-something hours of labor later, my wife was rushed backed to the operating room for an emergency c-section. Early on June 11, 2019 our precious baby boy made his grand entrance into this world. We did it! We were finally parents and man, was he perfect. William R.C. Krystallis was born a whopping 7lbs and 20inches long and in perfect health. Mommy and baby were doing great. My wife and I, despite being exhausted, refused to sleep. We talk, smiled, held and loved on our precious little boy.
William was such a great baby and was thriving. He was a natural at breastfeeding and hardly cried. Our nurse came in bright and early the morning after he was born to do some run of the mill testing on William. This would include a blood test to test for Cystic Fibrosis. The state we live in mandates that every newborn be tested for CF within 24 hours of birth. There was nothing to worry about. My wife had been tested during her pregnancy for the most common Cystic Fibrosis genes and was negative. Plus, William was very visibly healthy. Later that evening our doctor came in to tell us that my son’s CF blood work had come back slightly elevated. They tested his blood the next morning. Again, it came back just slightly elevated. We were told that they would have to do another test at his one week appointment. Multiple blood samples were taken over the next week. Each one coming back ever so slightly elevated. Although, not an alarming elevation for CF, each test was elevated enough to require more testing. After 4 total blood tests and several stool samples later, we were sent to a specialty lab to have what they call a sweat test performed on William. At this point, he was barely 2 weeks old. Our little guy had already been through so much and was such a trooper. As long as mommy was holding him he hardly made a peep during the tests. As we waited to hear back about his sweat test results, we had an appointment with a pediatric pulmonologist. While he was concerned that William’s tests were showing positive for Cystic Fibrosis, he assured us that the blood tests can sometimes show false positives but the final diagnosis would depend on the outcome of the sweat test.
A day shy of 3 weeks, my phone rang and the pediatric pulmonologist we had met with earlier in the week began to speak. He greeted me and within an instant, by the tone of his voice, I knew my life was about to change. “Mr. Krystallis, your son has Cystic Fibrosis.” That was all I heard. Everything else he said became murmurs in my now deaf ear. I held back the tears and interrupted the doctor. “I can’t tell her. You have to.” It was all I could manage to get out without crying. How was I supposed to walk down the hall, into my bedroom where my loving wife rocked our precious baby boy and tell her that the little baby she held had a potentially terminal illness? The baby boy that we wanted for so long, our perfectly healthy baby was very sick. I couldn’t do it. I couldn’t tell her. So, I walked down the hall and handed my wife the phone. I’ll never forget that moment. It was like a scene from a movie. She grabbed the phone, said hello and then took a deep breath. My wife slowly put the phone down, pulled our son to her chest and started crying. Time stood still. We spent the next several hours sitting on our bed, holding our precious and perfect little boy and crying. Crying because we knew the currently life expectancy for a person with cystic fibrosis was only 37 years. Our baby, that looked so healthy was actually sick and would only get sicker as he grew.
1 note
·
View note