
RSI- A leading regulatory Consultant in India provides all regulatory consulation that you need for cosmetic and medical device industry. Get in touch.
Don't wanna be here? Send us removal request.
Statistics
We looked inside some of the posts by regulatorysolutionsindia and here's what we found interesting.
Average Info
Notes Per Post
0
Likes Per Post
0
Reblog Per Post
0
Reply Per Post
0
Time Between Posts
20 days
Number of Posts By Type
Text
16
Video
1
Last Seen Tumblr Blogs





Fun Fact
In 2020, 27% of US Tumblr users had an annual household income of over $100,000.
Text
Exploring Medical Accessories: Understanding Accessories to Medical Devices in India
In the rapidly evolving Indian medical device industry, regulatory compliance is essential not just for primary devices but also for their integral components—accessories. In this article, we’re exploring medical accessories to understand their critical role in enhancing device functionality, patient safety, and legal compliance in India.
According to the Ministry of Health and Family Welfare’s notification S.O. 648(E) dated February 11, 2020, accessories to medical devices are considered medical devices themselves. This means exploring medical accessories from a regulatory standpoint is as important as understanding the primary devices they support. These accessories fall under the same legislative framework—the Drugs and Cosmetics Act, 1940, and the Medical Device Rules, 2017.
What Are Medical Accessories?
When exploring medical accessories, it's important to define them precisely. An accessory is a product intended by its manufacturer to be used with a specific medical device to enable, enhance, or support its intended function. They are not merely supplementary items; rather, they are critical for the performance and safety of parent devices.
For instance, exploring medical accessories like ECG electrodes or CPAP masks reveals that these are specifically designed to work with a primary system and ensure its optimal use.
Common Examples of Medical Accessories
Exploring medical accessories in practical scenarios involves understanding the breadth of items that qualify. Common examples include:
CPAP/BPAP nasal or face masks
SpO2 sensors
Blood pressure cuffs
Bone screws and plates for orthopedic implants
Pacemaker leads
Electrosurgical unit electrodes and cables
Cochlear implant magnets
X-ray cassettes and compatible printers
Nerve stimulator probes
Temperature probes
Surgical device adapters
Casting tapes and splint rolls
Trocar and cannula
Extension tubes
Y-connectors and stop cocks for infusion systems
Each of these is vital for delivering effective medical care and must be evaluated thoroughly when exploring medical accessories for compliance and functionality.
Regulatory Process for Accessory Registration
One of the most important aspects of exploring medical accessories is understanding how to register them. As per Notification S.O. 648(E), accessories are regulated as medical devices. Hence, the registration process mirrors that of standalone devices under the Medical Device Rules, 2017.
To comply:
Manufacturers and importers must register each accessory independently.
Accurate technical documentation, intended use rationale, and conformity assessments must be submitted.
Importers must also furnish regulatory clearances from their country of origin, where applicable.
A step-by-step guide is available in our comprehensive post: “A Guide to CDSCO Medical Device (incl. IVDs) Registration.”
Why Regulatory Expertise Matters

Exploring medical accessories also means navigating regulatory complexities. Certification involves a detailed understanding of product classifications, intended use statements, and supporting documentation. An experienced regulatory expert can streamline this process, ensuring your accessory meets CDSCO expectations and avoids delays or penalties.
How Regulatory Solutions India Supports You
Regulatory Solutions India (RSI), established in 2011, specializes in providing comprehensive regulatory services for medical devices and IVDs, including exploring medical accessories. Our services cover:
End-to-end support with CDSCO registration
Technical file preparation and dossier submission
Strategic regulatory planning for market entry in India
By exploring medical accessories with RSI, you gain a partner that understands India’s complex regulatory terrain and can facilitate timely, error-free approvals.
For those seeking to ensure compliance and streamline the market entry of accessories to medical devices in India, exploring medical accessories with the right knowledge and partners is the first crucial step.
Contact us today to begin your regulatory journey.
0 notes
Text
Understanding Accessories to Medical Devices in India
For the manufacture, import, and distribution of medical devices in India, companies must understand the regulatory procedures not only for the primary medical devices but also for the crucial components that support and enhance their functionality – the accessories. While accessories might seem like secondary items, they play a vital role in the effective and safe use of medical devices. Therefore, alongside the primary medical device, accessories are also regulated in India under the Drugs and Cosmetics Act, 1940, as notified through the S.O. 648 (E) dated February 11, 2020, issued by the Ministry of Health and Family Welfare. This signifies that accessories are subject to the same regulatory framework that applies to other medical devices in India.
To gain a deeper understanding of what constitutes accessories, how to register them, and other pertinent information, continue reading this blog.
What are accessories to medical devices?
An ‘Accessory’ means a device which can be added to a primary medical device to make it more useful, versatile, or attractive. According to the notification S.O. 648 (E), an accessory shall be specifically intended by its manufacturer to be used together (in combination) with a particular medical device (parent) to enable or assist that medical device (parent) to be used in accordance with its intended use. Therefore, it is considered a separate item, and manufacturers are required to register it independently.
What are the examples of accessories to medical devices?
Few examples of accessories to medical devices include:
CPAP/BPAP nasal/face mask
SpO2 sensor
Temperature Probes
Blood Pressure Cuffs
Adapters for surgical devices
X-ray cassettes
Bone screws or plates (used with implants)
Pacemaker leads
Electrosurgical unit electrodes and cables
Cochlear implant magnets
Trocar and cannula
3 way stop cock & Y-Connector for perfusion sets
Casting tapes/Splint Rolls
Printer for X-ray devices
Extension tubes
Nerve stimulator probes
How to register an accessory?
The process to register accessories to medical devices in India is same as that for medical devices themselves, as accessories fall under the definition of "medical devices" according to Notification S.O. 648(E) dated February 11, 2020. Therefore, they must be registered under the Drugs and Cosmetics Act, 1940, and the Medical Device Rules, 2017.
For a step-by-step guide on obtaining the registration certificate, check out our blog: “A Guide to CDSCO Medical Device (incl. IVDs) Registration”.
Why You Need an Expert to Navigate the Accessories Certification Process:
The Accessories Certification process often requires specialized knowledge of complex regulations. Therefore, an expert can efficiently guide you through the requirements, documentation, and submission stages, accelerating the process. This guidance can help you avoid costly mistakes and delays by ensuring accurate and thorough adherence to all requirements.
How Regulatory Solutions India Can Help You?
Regulatory Solutions India (RSI), established in 2011, specializes in providing end-to-end regulatory support to medical devices and IVDs manufacturers and importers worldwide. We assist in navigating the complex CDSCO regulatory landscape to ensure smooth registration and market entry of medical devices, IVDs, and their accessories in India. Our services include the preparation and submission of all required documentation, ensuring compliance with CDSCO standards.
With a deep understanding of Indian regulatory requirements, RSI can be your trusted partner in achieving timely and successful product registration and commercialization in the Indian market. Visit us to learn more.
0 notes
Text
Understanding Accessories to Medical Devices in India
0 notes
Text
Regulatory Pathway for In vitro diagnostics (IVD)
Registration in India: IVDs have become integral components of modern healthcare across the globe to support the diagnosis, monitoring, and personalized treatment of diseases. IVDs are also in very high demand in India, where the IVD market was worth about USD 2.64 billion in 2023 and expected to grow at a CAGR of 7.17% from 2024 until 2030 by Scania Analytics. As a result, the demand, interest, and enthusiasm of foreign IVD manufacturers to register and enter the Indian market have peaked.
However, before IVD devices can be introduced to India, manufacturers and importers must navigate a complex regulatory pathway. Moreover, understanding the registration process is crucial for entry into the Indian marketplace. This blog will help manufacturer and importers understand the IVD registration process in India, discussing the critical components necessary obtain a market launch in India. Keep reading for details.
What are IVD Devices?
IVD Devices (in vitro diagnostics) are instruments employed to analyze biological samples (e.g., blood, tissue, urine, or other bodily fluids) outside of the human body. These tests are integral in providing valuable indications for the diagnosis of disease, monitoring a patient’s condition, quantifying certain substances, and identifying infections. More familiar examples of in vitro diagnostic devices are pregnancy test kits, COVID-19 test kits, blood glucose monitoring kits, immunoassays, and human genetic testing devices.
Which Authority Regulates IVDs in India?
The authority in charge of licensing and regulating IVDS in India is the Central Drugs Standard Control Organization (CDSCO) under the Ministry of Health and Family Welfare.
How IVDs are classified?
IVDs are classified into four classes based on risk factors:
Class A: Low risk IVDs. Eg. Clinical chemistry analyzer (not near patient testing).
Class B: Low moderate risk IVDs. Eg. Pregnancy test strips, anti-nuclear antibody testing, urine test strips.
Class C: Moderate high risk IVDs. Eg. Blood glucose self-testing system, Human leukocyte antigen (HLA) typing, prostate-specific antigen (PSA) screening.
Class D: High risk IVDs. Eg. HIV blood donor screening, ABO/Rh(D) blood grouping analyser (near patient testing).
Central Licensing Authority’s Role in Regulating IVDs
The Central Licensing Authority is responsible for:
• Providing a license for the import of all classes of IVDs.
• Regulating the manufacture of Class C and Class D (high risk) IVDs.
• Providing the approval for clinical performance evaluation studies for new IVDs before they are allowed to be marketed in India.
• Working with State Licensing Authorities to ensure that there is consistent oversight of IVDs post-marketing.
Responsibilities of State Licensing Authority in Controlling IVDs
According to the Medical Devices Rules, 2017, the responsibilities of the State Licensing Authority are:
• To grant the licenses for the manufacture, sale or distribution of Class A or Class B (low risk) In Vitro Diagnostic medical devices in their state.
• To regulate the sale, stocking, exhibition and distribution of IVDs of all classes in their states.
How to register an IVD in India?

A manufacturer, importer or distributor can register an IVD in India by following these steps:
• Identify the Class of IVD: Identify the risk class for the device/personnel E or fQA Yo can more work.
• Designate the Authorized Indian Rep(s): Authorized agent if the applicant is a foreign manufacturer.
• Complete the Application Form: along with all supporting documents, for the developer or authorized agent on the SUGAM Portal:
Manufacturer: Applies for the Manufacturing license on Form MD-3orMD-4--Class A & B apply on Form MD-3 or MD-4. Class C and D manufacturers, MD-7 or MD-8.
More information about how to obtain a device manufacturing license can be read in our article, “5 Easy Steps To Obtain Your Medical Device Manufacturing License.”
Importers: Can apply for not only the manufacture device license but the device Import license using Form MD-14.The process of obtaining a licensing device can be perused at our blog, “Medical Devices Registration for Import.”
• Application for Review: The CDSCO will review the documents and may inspect overseas manufacturing. For example, review of importers data or evidence, while for manufacturers for India, will assign a State Licensing Authority (SLA)/ CLA a full site inspection process.
• License Issuance: Subject to all requirements being fulfilled, if the CDSCO is satisfied, a manufacturing or import license will be issued that is valid for five years unless otherwise noted.
How Regulatory Solutions India Can Help You?
If you have not registered your IVDs in India yet, RSI can assist you through the whole process. Having more than ten years in the medical device regulatory process, we have completed over 450 medical device registrations in all categories.
We have worked with clients in over 20 countries and can assist with everything from submission to regulatory approval in an effort to facilitate the approval process and secure your place in the market quicker. Let RSI help with gathering your IVD registration in India, contact us today!
#regulatory solutions india#regulatory consultancy in india#IVD#InVitroDiagnostics#MedicalDevices#FDA#RegulatoryAffairs#IVDR#MedTech#Biotech#HealthcareCompliance#QualityAssurance#MedicalDeviceRegulation#ClinicalDiagnostics#FDA510k#CEMarking#HealthcareInnovation
0 notes
Text
Regulatory Pathway for In Vitro Diagnostics (IVDs) | FDA 510(k), EU IVDR, Compliance Guide
Are you working in MedTech or regulatory affairs and need clarity on how In Vitro Diagnostics (IVDs) are approved and brought to market?
In this post (and video), we explain the entire regulatory pathway for IVDs, covering:
🔹 What are IVDs? 🔹 Classification of IVDs (Class A, B, C, D) 🔹 FDA 510(k), PMA, and EUA pathways 🔹 EU IVDR and CE marking 🔹 Clinical evaluation, labeling, and compliance
Whether you're a beginner, student, or experienced professional, this breakdown will help you stay compliant and competitive in the medical device industry.

#regulatory solutions india#regulatory consultancy in india#IVD#InVitroDiagnostics#MedicalDevices#FDA#RegulatoryAffairs#IVDR#MedTech#Biotech#HealthcareCompliance#QualityAssurance#MedicalDeviceRegulation#ClinicalDiagnostics#FDA510k#CEMarking#HealthcareInnovation
0 notes
Text
A Step-by-Step Guide to EPR Registration and EPR Certification in India
India's rapid industrial growth, fueled by increasing consumer demand, has led to a significant rise in environmental pollution, especially from plastic waste, e-waste, and batteries. To combat this issue, the Central Pollution Control Board (CPCB) and State Pollution Control Boards (SPCBs) have implemented Extended Producer Responsibility (EPR) regulations. These regulations make it mandatory for producers, importers, and brand owners (PIBOs) to manage the waste generated from their products.
EPR registration is now a legal requirement for businesses involved in plastic waste, e-waste, and battery management. Additionally, companies must implement waste management awareness programs. This guide will provide a step-by-step process for obtaining EPR registration and EPR certification in India.
What is Extended Producer Responsibility (EPR)?
Extended Producer Responsibility (EPR) is an environmental policy that holds Manufacturers, Importers, and Brand Owners (MIBOs) accountable for the post-consumer stage of their products' lifecycle. This includes waste collection, recycling, and proper disposal.
EPR regulations, notified by the Ministry of Environment, Forest, and Climate Change (MoEF&CC), are enforced by the CPCB and SPCBs. The aim is to reduce waste, promote recycling, and encourage sustainable product design. Businesses must complete EPR registration before selling products in India.
Who Requires EPR Registration?
EPR regulations apply to businesses engaged in the production, import, or usage of designated product categories, including:
Plastic packaging
Electrical and electronic equipment (EEE)
Batteries
For example, medical device manufacturers and importers dealing with plastic packaging, electrical/electronic components, or batteries must obtain EPR certification before distributing their products in India.
How to Register for EPR?
The EPR registration process involves the following steps:
1. Preparation of Documents
Compile the required documents, including a comprehensive EPR plan outlining the company’s commitment to environmental responsibility. A well-drafted EPR plan enhances the chances of approval.
2. Filling the Application
Once documents are ready, businesses must complete the EPR registration application in the prescribed format set by CPCB or SPCB. The application should be submitted along with the EPR plan and necessary supporting documents.
3. Payment of Fees
The EPR certification fee varies based on product category and scale of operations. The fee must be paid via the designated CPCB/SPCB payment gateway.
4. Scrutiny by Authorities
CPCB/SPCB will review the application, verify the provided details, and may request additional information if necessary.
5. Issuance of EPR Authorization
Upon satisfactory review, CPCB/SPCB will grant EPR registration, including a unique registration number and compliance conditions.
List of Medical Devices Requiring EPR Registration in India
The following medical devices require EPR certification:
Radiotherapy equipment and accessories
Cardiology equipment and accessories
Dialysis equipment and accessories
Pulmonary ventilators and accessories
Nuclear Medicine Equipment and accessories
Laboratory equipment for in vitro diagnosis and accessories
Analyzers and accessories
Magnetic Resonance Imaging (MRI), PET Scanner, CT Scanner, & Ultrasound Equipment
Fertilization test equipment and accessories
Other electronic medical devices used for diagnostics and treatment
EPR License Validity
EPR registration remains valid as per the waste category:
Plastic waste: Lifetime
Electronic waste: 5 years
Battery waste: 5 years
Benefits of EPR Registration

Obtaining EPR registration in India offers several benefits:
1. Legal Compliance
EPR ensures compliance with environmental regulations, helping businesses avoid legal penalties.
2. Improved Brand Reputation
Companies with EPR certification demonstrate environmental responsibility, boosting consumer trust and brand reputation.
3. Enhanced Waste Management
EPR promotes waste collection and recycling, leading to efficient resource utilization and reduced raw material costs.
4. Streamlined Business Operations
A structured waste management framework helps businesses optimize operations and reduce costs.
How Regulatory Solutions India Can Help
Regulatory Solutions India (RSI) has been a trusted partner for medical device companies since 2011. We provide expert assistance in EPR registration and EPR certification, ensuring compliance with CPCB/SPCB regulations. Our services include:
Complete documentation support
Seamless application process
Expert regulatory guidance
As one of the leading medical device regulatory consultants in India, we ensure your products meet all regulatory standards. Navigate the Indian medical device market with confidence. Contact us today to learn more!
0 notes
Text
A Step-by-Step Guide to EPR Registration (Plastics, E-Waste, Batteries) in India
#EPR Registration#epr certificate#Regulatory_Solutions_India#medical device import license registration
0 notes
Text
Requirements of Labeling of Medical Devices in India w.r.t. MDR 2017 & Amendments
In India, for the manufacture, import, and sale of medical devices, one must adhere to the Medical Device Rules, 2017, of the Central Drugs Standard Control Organization (CDSCO). The rules ensure that medical devices meet safety and quality standards. Labeling is one of the important elements in following the standards as it provides critical information to healthcare professionals and patients. The Legal Metrology Act, 2009, further requires that all pre-packaged goods, such as medical devices, comply with the prescribed labeling standards. It is essential to comply for both legal and transparency and accountability to consumers. This blog delves into the most important labeling requirements for medical devices under the Medical Device Rules, 2017, and why manufacturers and importers in India must strictly adhere to them. What is a Medical Device Label?A medical device label is an exhaustive list of all the vital information regarding the device, which includes its name, intended use, safety measures, manufacturer details, risks involved, precautions taken, expiry date, and many more, with the aim to safeguard the end-users and patients. Important Labeling Requirements of Medical Devices in India Medical devices, under the Medical Device Rules, 2017, are required to have certain labeling on the label, including but not limited to: Device Name: Clear identification of the medical device. Usage Details: The details that ensure users fully understand how and for what purpose the device is used. Manufacturer's Details: The manufacturer's name and address, and the address of the manufacturing location. Net Quantity: The exact quantity in terms of weight, volume or number of units, depending on the case. Date of Manufacture/Expiry: The month and year date of manufacture and expiration or shelf life of the device. Sterilization Information: For items that are sterile, date sterilized and method. Biological/Medicinal Product: If the item contains a medicinal or biological product. Storage/Handling Instructions: Other specific storage and handling instructions. Warnings/Precautions: Any caution or information related to hazards. Batch or Lot Number: Traceability in form of batch or lot number. UDI: As required by Central Government Single-Use Devices: Mark single-use devices as such. Regulatory Numbers: Manufacturing license number (for domestic devices) or import license number and details for imported devices. Additionally, labels may include recognized symbols from the Bureau of Indian Standards or ISO, to provide further clarity and standardization. Why is Medical Device Labeling Crucial?

Patient Safety: Proper labeling ensures safe usage, reducing the risks of misuse or adverse events. Regulatory Compliance: Failure to comply with labeling standards may result in penalties, recalls or market withdrawal. Traceability: The labels have batch and lot numbers and UDI to help track devices at the time of defects. Risk Management: Labels contain warnings and precautions to reduce potential risks. Storage & Handling: Proper guidelines for maintaining and storage keep the devices working.
How Regulatory Solutions India Can Help You? Regulatory Solutions India is an affiliation that has been a reliable partner to a host of medical device manufacturers and importers since 2011. The leading role in the CDSCO labeling regulations can guide you all through preparing, submitting, and ensuring labels meet the right criteria to get to approval. Partner with RSI to navigate the complex labeling requirements for the medical devices in India, ensuring compliance, safety, and success in the market. Visit our blog on Legal Metrology Compliance for Medical Devices in India to know more about labeling and compliance.
#labeling_requirements#medical_devices_labeling_requirements#labeling_requirements_of_medical_devices#regulatory_solutions_india
0 notes
Text
How Regulatory Reporting Can Help in Medical Device Introduction in India
The Indian medical device market is growing at an unprecedented rate. Currently valued at approximately USD 12 billion, it is projected to reach USD 50 billion by 2030. This growth presents a lucrative opportunity for manufacturers and importers to introduce their medical devices in India.
However, entering this market requires careful navigation through stringent regulatory approval processes. To ensure a successful launch, having a robust plan for regulatory reporting is crucial.
In this blog, we will explain how regulatory reporting can assist in introducing a medical device in the Indian market.
Governing Body for Medical Device Registration in India
The Central Drugs Standard Control Organization (CDSCO) serves as the National Regulatory Authority of India, overseeing medical device registration and ensuring compliance with regulatory standards.
Who Can Apply for Medical Device Registration Under CDSCO?
The following parties are eligible to register medical devices with CDSCO:
Manufacturers with a registered office in India
Authorized representatives of the manufacturer
Subsidiaries of the manufacturer
Importers
Domestic manufacturers
For more information, refer to our blog, "7 Key Steps In CDSCO Medical Device Registration: Easy Guide."
What is Regulatory Reporting?
Regulatory reporting involves the systematic preparation and submission of documentation required for gaining approval to launch medical devices in the Indian market. This process is key to ensuring compliance with CDSCO regulations and other local standards.
Importance of Regulatory Reporting
Effective regulatory reporting facilitates:
Timely Market Access: Accelerating product approvals for quicker entry into the market.
Risk Minimization: Identifying and mitigating potential regulatory risks.
Regulatory Compliance: Ensuring adherence to evolving standards and regulations.
Resource Optimization: Enabling efficient allocation of resources during the approval process.
Key Components of Regulatory Reporting
A comprehensive regulatory reporting strategy consists of the following elements:
Product Type Identification Determine whether the product falls under medical devices, In Vitro Diagnostics (IVDs), cosmetics, or drugs, as each has distinct regulatory frameworks.
Risk Classification Establish the risk class of the device, as this impacts the regulatory requirements and approval pathway.
Existing Predicate or Similar Device Identify if a similar device exists in India. A predicate device demonstrates substantial equivalence and simplifies the approval process. If no predicate exists, additional steps are required. For more details, refer to our blog, "CDSCO Approval Process for Medical Devices Without Predicate in India."
Regulatory Status in Other Countries Leverage approvals from other countries to demonstrate safety, efficacy, and quality. This may also qualify the device for certain waivers, such as clinical trial exemptions.
Warehouse Availability & Registration Ensure that a warehouse is available and registered (Form MD-42). This is a mandatory prerequisite for introducing a medical device in India. Refer to our blog, "Registration Certificate for Sale," for further guidance.

Conclusion
The Indian medical device market presents tremendous opportunities but also demands adherence to stringent regulatory requirements. Comprehensive regulatory reporting ensures a clear pathway for product approval, minimizing delays and ensuring compliance.
Regulatory Solutions India (RSI) offers over 12 years of expertise in regulatory reporting, guiding companies through the complex process of medical device, IVD, cosmetics, and drug registration. Contact us today to streamline your product’s journey into India’s rapidly growing healthcare market. Partnering with RSI will help you bring your innovations to patients while ensuring compliance and commercial success.
0 notes
Text
How Regulatory Reporting Can Help in Medical Device Introduction in India
The Indian medical device market is growing at an unprecedented rate. Currently valued at approximately USD 12 billion, it is projected to reach USD 50 billion by 2030. This growth presents a lucrative opportunity for manufacturers and importers to introduce their medical devices in India.
However, entering this market requires careful navigation through stringent regulatory approval processes. To ensure a successful launch, having a robust plan for regulatory reporting is crucial.
In this blog, we will explain how regulatory reporting can assist in introducing a medical device in the Indian market.
Governing Body for Medical Device Registration in India
The Central Drugs Standard Control Organization (CDSCO) serves as the National Regulatory Authority of India, overseeing medical device registration and ensuring compliance with regulatory standards.
Who Can Apply for Medical Device Registration Under CDSCO?
The following parties are eligible to register medical devices with CDSCO:
Manufacturers with a registered office in India
Authorized representatives of the manufacturer
Subsidiaries of the manufacturer
Importers
Domestic manufacturers
For more information, refer to our blog, "7 Key Steps In CDSCO Medical Device Registration: Easy Guide."
What is Regulatory Reporting?
Regulatory reporting involves the systematic preparation and submission of documentation required for gaining approval to launch medical devices in the Indian market. This process is key to ensuring compliance with CDSCO regulations and other local standards.
Importance of Regulatory Reporting
Effective regulatory reporting facilitates:
Timely Market Access: Accelerating product approvals for quicker entry into the market.
Risk Minimization: Identifying and mitigating potential regulatory risks.
Regulatory Compliance: Ensuring adherence to evolving standards and regulations.
Resource Optimization: Enabling efficient allocation of resources during the approval process.
Key Components of Regulatory Reporting
A comprehensive regulatory reporting strategy consists of the following elements:

Product Type Identification Determine whether the product falls under medical devices, In Vitro Diagnostics (IVDs), cosmetics, or drugs, as each has distinct regulatory frameworks.
Risk Classification Establish the risk class of the device, as this impacts the regulatory requirements and approval pathway.
Existing Predicate or Similar Device Identify if a similar device exists in India. A predicate device demonstrates substantial equivalence and simplifies the approval process. If no predicate exists, additional steps are required. For more details, refer to our blog, "CDSCO Approval Process for Medical Devices Without Predicate in India."
Regulatory Status in Other Countries Leverage approvals from other countries to demonstrate safety, efficacy, and quality. This may also qualify the device for certain waivers, such as clinical trial exemptions.
Warehouse Availability & Registration Ensure that a warehouse is available and registered (Form MD-42). This is a mandatory prerequisite for introducing a medical device in India. Refer to our blog, "Registration Certificate for Sale," for further guidance.
Conclusion
The Indian medical device market presents tremendous opportunities but also demands adherence to stringent regulatory requirements. Comprehensive regulatory reporting ensures a clear pathway for product approval, minimizing delays and ensuring compliance.
Regulatory Solutions India (RSI) offers over 12 years of expertise in regulatory reporting, guiding companies through the complex process of medical device, IVD, cosmetics, and drug registration. Contact us today to streamline your product’s journey into India’s rapidly growing healthcare market. Partnering with RSI will help you bring your innovations to patients while ensuring compliance and commercial success.
0 notes
Text
Mastering Legal Metrology Compliance for Medical Devices: Your Complete Guide
The manufacturing, sale, and distribution of medical devices are governed by strict regulations to ensure their safety and efficacy. Beyond registering devices with relevant authorities, manufacturers and importers must adhere to the Legal Metrology (Packaged Commodities) Rules, 2011. These rules are essential for maintaining transparency, protecting consumers, and fostering fair trade practices.
This guide unpacks the key provisions of Legal Metrology compliance and explains how adhering to these rules can enhance your business's credibility and operational efficiency.
What Are the Legal Metrology (Packaged Commodities) Rules, 2011?
The Legal Metrology (Packaged Commodities) Rules, 2011, established under the Legal Metrology Act, 2009, regulate the labeling and packaging of pre-packaged goods, including medical devices. These regulations ensure that consumers receive accurate, clear, and detailed information about the products they purchase. Compliance fosters consumer trust, prevents misleading claims, and upholds ethical business practices.
Key Provisions of Legal Metrology Rules
Labeling Requirements
Each packaged commodity must include a label with the following mandatory details:
Name and address of the manufacturer, packer, or importer.
Common or generic name of the product.
Net quantity in standard units (weight, measure, or number).
Manufacturing, pre-packing, or import date (month and year).
Retail sale price (MRP).
Dimensions or quantity of the commodity.
Display Panel Requirements
All declarations must appear on the principal display panel.
Information must be legible and prominent.
Net quantity and price should be printed in contrasting colors.
Labels must be in Hindi or English.
Wholesaler and Retailer Obligations
Wholesalers and retailers cannot sell, store, or display packaged commodities unless they meet all Legal Metrology provisions.
Retailers must not sell packaged products at prices exceeding the declared MRP.
Why Legal Metrology Compliance Matters
Fair Trade and Consumer Protection Transparent labeling ensures that consumers can make informed purchasing decisions.
Avoidance of Penalties Non-compliance can lead to fines, legal actions, or operational disruptions.
Market Transparency Accurate declarations enhance accountability and build consumer trust.
Efficient Inventory Management Compliance facilitates better inventory tracking and streamlined sales processes.
How to Obtain a Legal Metrology Packaged Commodities (LMPC) Certificate
To comply with the Legal Metrology Rules, manufacturers and importers must obtain an LMPC Certificate. Here’s the process:
Registration Apply under Rule 27 of the Legal Metrology (Packaged Commodities) Rules, 2011.
Document Submission Submit required documents, including business details, product information, and sample labels.
Review and Approval Authorities review the documents and grant the LMPC Certificate upon successful compliance.
Why Seek Expert Guidance for Legal Metrology Compliance?
Navigating the Legal Metrology certification process can be complex, involving technical and legal intricacies. Engaging a professional consultancy can save time, ensure compliance, and minimize errors, helping you focus on your core business.

How Regulatory Solutions Can Help
Regulatory Solutions has been a trusted partner for manufacturers and importers for over a decade, ensuring seamless compliance with regulatory frameworks, including the Legal Metrology (Packaged Commodities) Rules, 2011.
We offer:
Expertise in medical device, IVD, and cosmetic registrations.
A proven track record with a diverse range of products and categories.
Comprehensive support in obtaining the LMPC Certificate.
Ensure your products meet regulatory standards and build trust with your consumers. Contact us today to learn more about how we can support your compliance journey.
#legal metrology registration#legal metrology act#legal metrology rules#LPMC#Legal Metrology Packaged Commodities Rules
0 notes
Text
A Guide to Legal Metrology Compliance for Medical Devices in India
If you're working with medical devices in India, it's crucial to understand the Legal Metrology Act and its compliance requirements. This guide covers everything from labeling and packaging standards to the necessary regulations for medical device manufacturers, importers, and distributors.
🔑 Key Points:
Overview of the Legal Metrology Act and its impact on medical devices.
Compliance requirements for labeling, packaging, and more.
Tips for navigating common regulatory challenges.
Stay informed and compliant to avoid legal issues and ensure your medical devices meet the required standards. 💼⚖️
#LegalMetrology#MedicalDevicesIndia#Compliance#MedicalDeviceRegulations#IndiaBusiness#LegalCompliance#HealthcareRegulations#PackagingStandards#Labeling#MetrologyCompliance
0 notes
Video
youtube
In this video, we break down the essential aspects of Legal Metrology Compliance for medical devices in India. Learn about the regulations, standards, and requirements that manufacturers, importers, and distributors must follow to ensure compliance with the Legal Metrology Act. We also explore key insights into labeling, packaging, and other critical areas for medical device businesses. Stay updated and compliant to avoid legal complications and ensure smooth operations.
🔹 Key topics covered:
Overview of Legal Metrology Act
Compliance requirements for medical devices
Labeling and packaging standards
Key challenges and how to navigate them
Subscribe for more updates on regulations and compliance in the medical device industry!
0 notes
Text
Unlocking Compliance: Essential Guide to the Non-Conviction Certificate for Medical Devices in India
In India, medical device registration is essential for manufacturers, importers, and distributors aiming to ensure regulatory compliance and public safety. Governed by the Central Drugs Standard Control Organization (CDSCO) under the Medical Device Rules 2017, the process requires various approvals, including CDSCO medical device registration, product approvals, import licenses, and the Non-Conviction Certificate (NCC). Among these, the NCC is a crucial document that reinforces a company’s compliance record. This guide delves into the Non-Conviction Certificate, highlighting its importance, eligibility, and the steps involved in obtaining it.
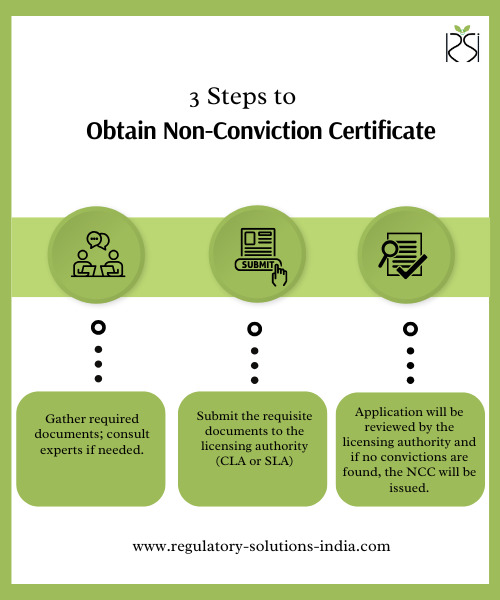
What is a Non-Conviction Certificate (NCC)?
A Non-Conviction Certificate (NCC) is issued by the CDSCO to Indian medical device companies. This certificate attests that a company has not been convicted of offenses related to safety, quality standards, or product malfunctions associated with its devices. Often essential in international tenders and product registration in foreign markets, the NCC demonstrates a company's adherence to high regulatory standards. This certification is vital in the medical device regulatory services landscape in India, underscoring the company’s commitment to ethical practices.
Importance of the Non-Conviction Certificate (NCC)
The Non-Conviction Certificate has numerous advantages for medical device companies, including:
- Building Trust: Demonstrates a company’s dedication to safety, quality, and regulatory compliance. - Unlocking Business Opportunities: Required in many tenders and pre-qualification applications, especially in CDSCO medical device registration. - Maintaining Market Reputation: Strengthens a company’s credibility and reputation in the medical devices India sector.
Eligibility Criteria for the Non-Conviction Certificate
To be eligible for the NCC, a company must: - Hold a valid medical device registration India license from the Central Licensing Authority (CLA) or State Licensing Authority (SLA). - Maintain a clean record with no prior convictions or violations under the Drugs and Cosmetics Act 1940 and related Rules.
Required Documentation for the Non-Conviction Certificate
Obtaining an NCC involves compiling the following documents: 1. A cover letter stating the purpose of the application. 2. A copy of the company's valid medical device registration or import license. 3. A list of products for which the NCC is being requested. 4. Prescribed government fees. 5. A Legal Undertaking on a ₹100 registered, notarized stamp paper, confirming no convictions related to product malfunctions or compliance violations.
Who Issues the Non-Conviction Certificate?
The licensing authority that initially issued the manufacturing or import license (either the CLA or SLA) is responsible for granting the NCC.
Validity of the Non-Conviction Certificate
The NCC is valid for one year from issuance or until the company's manufacturing license expires, whichever occurs first.
Steps to Obtain the Non-Conviction Certificate
1. Submit the application with the necessary documents to the relevant licensing authority. 2. For a smooth application process, consider engaging a regulatory consultant in India who specializes in medical device regulatory consultancy.
Benefits of Hiring a Regulatory Consultant for NCC Application
Engaging a medical device regulatory consultant in India can ease the application process, given their expertise in regulatory consultancy services. Consultants bring several advantages: - Requirement Clarity: Ensures the applicant meets eligibility criteria and provides complete documentation. - Efficient Navigation of Procedures: Streamlines applications and reduces potential delays. - Expert Advice: Consultants bring in-depth knowledge, particularly beneficial for medical device registration India requirements. - Peace of Mind: Consultants manage the regulatory process, allowing companies to focus on core operations.
Consult Regulatory Solutions India for Your NCC Needs
Regulatory Solutions India (RSI) offers expert regulatory consultancy services for medical devices and IVDs in India, providing end-to-end support for compliance, including CDSCO consultancy services. With RSI's guidance, you can ensure compliance with regulatory standards, obtain the Non-Conviction Certificate, and gain a competitive edge in India’s regulated markets. ContactRSI today to discuss your specific requirements. Our team of regulatory consultants is here to assist with your CDSCO medical device registration and NCC needs, making the compliance journey smoother and more efficient.
0 notes
Text
Unveiling the Future of India’s Medical Devices Market: Opportunities and Challenges
In the past decade, the medical devices market in India has witnessed a remarkable surge, driven by growing demand for various medical devices, including consumables, dental products, diagnostic imaging equipment, prosthetics, orthopedics, patient aids, and more. To support and regulate this booming sector, the government has enacted several policies and reforms. These initiatives aim to foster the development of new technologies, ensure healthcare safety and quality, and meet the diverse needs of India’s health system.
However, despite these advancements, a significant gap remains between the demand for medical devices in India and their availability. To address this, the government has made notable changes to the Foreign Direct Investment (FDI) policy, encouraging the importation of medical devices to bridge the supply gap.
In this blog, we will explore the medical devices market in India and discuss how foreign medical device companies can successfully enter the Indian marketplace.
Market Volume of Medical Devices in India
India stands as the fourth-largest market for medical devices in Asia, behind Japan, China, and South Korea, and is among the top 20 global markets for medical devices. Let’s dive into the numbers that illustrate the size of India’s medical devices sector:
- In 2022, the medical devices market in India was valued at approximately Rs. 90,000 million (about US$11 billion). The market is projected to grow to US$50 billion by 2030, with a compound annual growth rate (CAGR) of 16.4%.
- The diagnostic equipment segment is expected to reach US$6 billion by 2027, with a CAGR of 16.4% from 2020 to 2030.
- India’s reliance on imported medical devices grew by 21% between November 2022 and October 2023, with imports totaling Rs. 61,262.84 crore (US$7.23 billion).
Key Drivers of Growth in the Medical Devices Market in India

Several factors contribute to the rapid growth of the medical devices market in India. These include:
- Growing Population: With a population of over 1.4 billion, there is rising demand for medical devices to address chronic diseases, advanced diagnostics, and treatment options.
- Aging Demographics: By 2030, India’s elderly population is expected to reach 194 million, driving demand for medical devices that cater to mobility, diagnostics, and chronic disease management.
- Increase in Chronic Diseases: According to the International Diabetes Federation, India’s diabetes prevalence is predicted to reach 74% by 2025. Alongside this, the rise of other chronic conditions such as cancer and cardiovascular diseases further fuels the need for advanced diagnostic and therapeutic devices.
- Expansion of Health Insurance Coverage: Government initiatives like Ayushman Bharat, which provides health benefits to 500 million citizens, have significantly increased the need for medical devices in India.
- Rising Middle Class and Health Awareness: By 2026, 8% of Indians are expected to earn over USD 12,000 annually, and 73 million households will join the middle class within the next decade. This economic growth and awareness are driving healthcare spending and the adoption of health technologies.
- Growth of Private Healthcare: India’s private healthcare sector has seen rapid expansion, with 393 hospitals accredited by the National Accreditation Board for Hospitals & Healthcare Providers (NABH) by 2019, increasing demand for high-quality medical devices.
- Medical Tourism: India is becoming a major hub for medical tourism, attracting nearly 2 million international patients annually and generating US$6 billion in revenue, projected to reach US$13 billion by 2026. The medical devices market in India is a crucial enabler of this growth, supported by the government’s “Heal in India” initiative.
- Favorable Regulatory Environment: Regulatory reforms, such as the Medical Devices Rules, 2017, have streamlined access to the Indian market, supporting both domestic manufacturers and importers.
How Foreign Medical Device Companies Can Access the Indian Market
Foreign companies looking to enter the medical devices market in India should follow these steps:
1. Market Research: Conduct thorough research on market demand, pricing, competition, and regulatory requirements to identify export opportunities.
2. Regulatory Compliance: Familiarize yourself with India’s regulatory landscape, especially the Central Drugs Standard Control Organization (CDSCO), which governs medical device registration and approval.
3. Leverage Government Initiatives: Take advantage of tax benefits, regulatory harmonization efforts, and policies promoting high-quality medical devices in India.
4. Quality Assurance: Ensure compliance with the Bureau of Indian Standards (BIS), ISO, and IEC guidelines to build trust among Indian consumers.
5. Regulatory Support: Partner with regulatory consultants specializing in the medical devices market in India to navigate the approval process and ensure successful entry.
Future Outlook
With continued government support and technological advancements, the medical devices market in India is poised for robust growth. The government’s focus on regulatory ease, fostering innovation, and creating a favorable manufacturing environment will facilitate greater market access for foreign manufacturers, helping them capitalize on India’s rapidly expanding healthcare sector.
At Regulatory Solutions India (RSI), we specialize in regulatory consulting for medical devices, IVDs, cosmetics, and more. Let us help you navigate the medical devices market in India and achieve success. Send us an email here to discuss your specific needs and we’ll move forward from there.
Recent Blogs
Insight into the Medical Devices Market in India
Registration of Cosmetics in India
Materiovigilance Programme of India (MvPI) :DCGI Calls for Strengthening Medical Device Adverse Event Reporting
Intraocular Lenses and Regulatory Processes
CDSCO Update: New Online PSUR Guidelines in India
CDSCO Test License for Medical Devices in India
NSWS portal: A Single Window for Central Government Approvals
CDSCO Approval Process for Medical Devices without predicate in India
Ensuring Quality in Healthcare: The Role of BIS Standards for Medical Devices
6-Month Extension Announced for Class C and Class D Medical devices
#medical devices market in India#BIS Certification for Medical Devices#CDSCO Medical Device Import License#Medical Devices Market
0 notes
Text
Registration of Cosmetics in India
Cosmetics are utilized to improve a person’s appearance. These are used for various beauty treatments such as skin tightening, hair removal, spot reduction, achieving radiant skin, and many more. They play a critical role in boosting an individual’s self-confidence and positive outlook. Consequently, there has been a significant increase in demand for cosmetics in the Indian market, resulting in substantial growth in the cosmetic industry in recent years. However, ensuring the highest quality and safety of cosmetics remains a major concern for the industry.
For this reason, it is mandatory to register every cosmetic in India. The registration process must be compliant with the Drugs and Cosmetics Act of 1940 and the Cosmetic Rules of 2020. The Central Drug Standard Control Organisation (CDSCO), under the Ministry of Health and Family Welfare, is the regulatory authority responsible for overseeing these regulations. All cosmetics manufactured in or imported into India must be registered with the CDSCO.
Definition of Cosmetics as per the Drugs and Cosmetics Act, 1940
Under section 3(aaa) of the Drugs and Cosmetics Act, 1940, cosmetics is defined as, “any article intended to be rubbed, poured, sprinkled or sprayed on, or introduced into, or otherwise applied to, the human body or any part thereof for cleansing, beautifying, promoting attractiveness or altering the appearance, and includes any article intended for use as a component of cosmetic”.
Under the provisions of the aforesaid Act, the manufacture of cosmetics is regulated by the State Licensing Authorities appointed by the respective State Governments, while the import of cosmetics is regulated by the Central Licensing Authority appointed by the Central Government. The Drugs Controller General (India) is the Central Licensing Authority who grants registration certificate for import.
Key Requirements for Cosmetics in India
To ensure the safety, quality, and efficacy of cosmetics in India, key requirements under the Cosmetic Rules, 2020 are as follows:
All cosmetics manufactured in or imported to India must comply with the Cosmetic Rules, 2020.
All manufacturers must obtain a license or loan license from the State Licensing Authority to manufacture cosmetics for sale and distribution in India.
All importers must obtain an import registration certificate from the Central Licensing Authority to import cosmetics to India.
All the manufacturers of cosmetics in India must label and pack the cosmetics in accordance with the Cosmetic Rules, 2020 and Legal Metrology (Packaged Commodities) Rules, 2011, before selling or distributing the product.
Additional Regulatory Requirements for Cosmetics in India
Cosmetics should not contain any of the raw materials listed in Indian Standard IS: 4707.
Cosmetic products should not contain dyes, colours, or pigments other than those specified by the Bureau of Indian Standards (IS: 4707).
Cosmetic products that contain permitted synthetic organic and natural organic colours should not contain arsenic trioxide, lead, mercury, or heavy metals in excess of the quantities specified in the Cosmetic Rules, 2020.
Hexachlorophene should not be an ingredient in any cosmetic.
Manufacturers should not use animals for testing cosmetics.
Process to get Import Registration Certificate
Under Sections 12 and 13 of the Cosmetic Rules, 2020, a foreign manufacturer's authorised agent or authorised subsidiary may obtain import registration certification through the following process:
Apply to register cosmetics intended for import into India through the central government's online portal, Form COS-1.
The Form COS-1 can be submitted either by the manufacturer himself or his authorized agent or the importer or an Indian subsidiary authorized by the manufacturer.
If the Central Licensing Authority deems the documents provided with the application satisfactory, it may grant the applicant the Import Registration Certificate. The Central Licensing Authority may also reject an application, documenting its reasons in writing within six months of the application date.
If the Central Licensing Authority rejects the application, the applicant has forty-five days to appeal to the Central Government. If the government considers it necessary, it can pass orders in relation thereto within a period of ninety days from the date of appeal.
Before registering the import of a new cosmetic into India, the applicant must obtain prior permission from the Central Licensing Authority in Form COS-3 before registration of the cosmetic.
Process to get Licence or Loan Licence to Manufacture Cosmetics for Sale or Distribution
Under Section 23 of the Cosmetic Rules, 2020, anyone intending to manufacture cosmetics for sales and distribution should obtain a license from the State Licensing Authority through the following process:
Apply for a license through an identified online portal, (can apply offline if online portal is not operational) in Form COS-5 for a license or in Form COS-6 for a loan license.
For a new cosmetic, the manufacturer must obtain prior approval in Form COS-3 from the Central Licensing Authority.
In addition to the required documents, the applicant must also submit a self-declaration in Form COS-7 conforming to Good Manufacturing Practices and additional manufacturing related requirements.
Upon receipt of the application, within a period of forty-five days, the State Licensing Authority will grant a license or loan license after confirming that all requirements have been met or will inform the applicant if it determines that the applicant has not fulfilled the requirement.
Within thirty days from the date of grant of the license or loan license, the manufacturing site will be inspected by the subordinate officer delegated by the State Licensing Authority to verify the information given in the self-certificate in Form COS-7.
Requirements for Registration of Cosmetics for Import
Following is the list of main documents/details that need to be submitted at the time of applying for a cosmetic registration for import.
Authorization from Manufacturer as per First Schedule
Product details and undertaking as per Second Schedule Part I
Regulatory Certificates (manufacturing license/Free Sale Certificate)
Non-Animal Testing Declaration
Declaration for Heavy Metal and Hexachlorophene content
Applicable Government Fees to be paid

Conclusion
The regulations for registration and import of cosmetics in India are crucial for ensuring the quality of cosmetics and safeguarding the well-being of the consumers. Therefore, any manufacturer or importer/authorized agent involved in the cosmetics industry must follow these regulations to ensure the quality and safety of all.
At Regulatory Solution India (RSI), we specialize in providing regulatory consulting services for cosmetics. If you need assistance navigating the submission process or ensuring compliance with the latest regulations, Contact us.
#Drugs and Cosmetics Act#Registration of Cosmetics in India#Cosmetics#Requirements for Registration of Cosmetics for Import
0 notes
Text
Materiovigilance Programme of India (MvPI): DCGI Calls for Strengthening Medical Device Adverse Event Reporting
The Drugs Controller General of India (DCGI) has called for the timely reporting of adverse events related to medical devices by issuing a circular dated May 15, 2024. The move aims to strengthen the existing Materiovigilance Programme of India (MvPI) ensuring that medical devices are safe and reliable once in the market.
What does the Circular state mean?
The Materiovigilance Programme of India (MvPI) is an important programme for reporting adverse events, coordinated analysis related to medical devices, including in-vitro diagnostic devices, therefore it is suggested that all the license holders should also use the MvPI platform to report any adverse events/serious adverse events associated with the devices to enhance the procedure of identifying risk associated with medical devices.
What is MvPI? The Materiovigilance Programme of India (MvPI) was launched by the Ministry of Health and Family Welfare, Government of India, on July 6, 2015, with the objective to monitor, record and analyse the root cause of adverse events or risks associated with the use of medical devices, including in-vitro diagnostics by healthcare professionals or patients/users and suggesting regulatory bodies to take appropriate action to improve Indian patients’ safety. Indian Pharmacopeia Commission (IPC) is functioning as National Coordination Center (NCC) for MvPI.
Objectives of MvPI:
The core objectives of MvPI are as follows:
· To promote the reporting of adverse events related to medical devices by clinicians, biomedical engineers/clinical engineers, hospital technology managers, pharmacists, nurses, technicians, medical-device manufacturers.
· To collect adverse event reports, assess them, and submit medical device reports to the medical device regulator.
· Voluntary registration of medical device manufacturers to:
· Report adverse events, associated.
· Root cause analysis and corrective/prevention action to IPC-NCC.
· To develop and implement an electronic reporting system (e-reporting).
· To support the health system where in procurement of medical device is only undertaken after studying adverse events associated with the medical device intended for procurement.
· To make it mandatory, in the long term, — for all healthcare providers under the Clinical Establishment Act to report adverse events associated with medical devices.
Important Responsibilities under MvPI:
Medical device organisations, including in-vitro diagnostics, are important stakeholders to make the MvPI programme a success. The key responsibilities of such organisations as a result of the MvPI are as follows:
· Robust Post-Market Surveillance: Post-market surveillance refers to the vigilance on the use of medical devices, including the information on the quality, safety. or performance of medical devices after the medical device is placed on the market. With a robust post-market surveillance system, organizations can reduce, -the chances of adverse events that may lead to any harm/ injury to the patient. Moreover, the regular vigilance process reduces the likelihood of the same type of adverse incident being repeated in different places at different times.
· Reporting of Medical Device Adverse Event: In case an adverse event or incident related to a medical device has been noticed by the manufacturer or healthcare service provider, or Medical Device Adverse Event Monitoring Centers (of public/private hospitals), it should be reported immediately, and its root cause of failure should be investigated and intimated to IPC-NCC.
· Reporting of Field Safety Corrective Action: A field safety corrective action (FSCA) notification form has been developed to notify the regulatory authority and the consignees regarding any corrective action or recall that has been initiated by the manufacturer/importer to reduce any serious adverse reaction associated with the use of medical devices. Concerned organizations must take cognizance of this form and use it for reporting their FSCAs.

Benefits of MvPI:
It is crucial for providing secured, sensible and responsible healthcare to citizens in India.
It contributes to establishing a system in India for systematic, scientific and practical means of screening large medical device adverse events datasets at the national level.
It contributes to public health by identifying potential safety issues more accurately and quickly.
It promotes patient safety by identifying adverse events that could result in patient harm.
It encourages Medical Device Manufacturers/ Importers to put products in market with a sense of ethical business, analyze and improve design and performance of products.
It can provide supportive data to improvise product standards developed by ISO/BIS.
Conclusion:
MvPI is a critical components of India's healthcare delivery system, guaranteeing the safety and efficacy of medical devices while increasing healthcare quality. Prioritizing transparency and employing cutting edge technology can create an efficient Materiovigilance system which can protect public health while improving healthcare quality. For more information on India's Materiovigilance Programme and its effect on medical device safety, visit, https://www.ipc.gov.in/mandates/materiovigilance-programme-of-india-mvpi/about-us.html.
How can Regulatory Solutions India Help You?
Regulatory Solutions India (RSI) is your reliable partner when it comes to medical device regulation. Since 2011, they have successfully registered over 450 products ranging from medical devices, IVDs and cosmetics across 25+ categories such as stents, catheters, intraocular lenses, orthopedic implants, ablation devices surgical dressings, hypodermic syringes/needles and more from clients from 15+ countries. RSI provides its clients with a unique combination of technical, strategic and project management support backed by rich industry experience. We help companies register their products with India's central licensing authority (CDSCO) while offering expert knowledge into CDSCO medical device registration and import license processes in order to navigate successfully the regulatory landscape.

Why Choose RSI?
Regulatory Strategy for Medical devices: Analyse the portfolio, interpret regulatory requirements, identify potential problems, and design the right strategy for medical devices accordingly.
Regulatory Application Submission to CDSCO: Review regulatory documents, validate dossiers and submit applications for Import License.
Registration Approval: Technical support for scientific meetings with regulators and for responding to regulatory queries.
Post Registration Support: It also offers post registration support in labeling recommendations & regulatory compliance, post registration compliance reporting obligations and advise on impact, if any, due to regulatory changes
Take Action Today
Partner with Regulatory Solutions India to ensure the safety and security of the medical devices. Visit us here.
0 notes