
Last Seen Blogs


sbautosounds
Auto Sounds Car Stereo & Window Tinting SB

blazas
Blazas jungle

livingthesuperherolife
Living the Superhero Life

andrisutrisna-blog
ANDRI SUTRISNA
Text
me: say it— i need to hear those three words
library database: Full Text Online
me, shedding tears: i love you too
312K notes
·
View notes
Text
Things almost every author needs to research
How bodies decompose
Wilderness survival skills
Mob mentality
Other cultures
What it takes for a human to die in a given situation
Common tropes in your genre
Average weather for your setting
488K notes
·
View notes

Photo

Mitochondria found to run as high as 50 C
A team of researchers with members from France, Korea and Germany has found that temperatures inside human mitochondria can run as high as 50°C. In their paper uploaded to the preprint server bioRxiv, the group describes how they used temperature-sensitive dyes to determine the temperature of the organelles and what their findings might mean for prior biological research that has relied on lower temperatures during reactions.
The healthy human body is supposed to maintain a constant temperature of 37°C, but now it appears our cells may actually run much hotter. In this new effort, the researchers took the temperature of mitochondria in skin, kidney and lung cancer cells, and found them to be much hotter than was anticipated.
Because it is impossible with existing technology to take the temperature of mitochondria using conventional thermometers, the team took a new approach, applying a type of dye that changes color depending on the temperature of its environment. The dye, called Mito-Thermo-Yellow, binds to targets, and loses fluorescence as the temperature around it rises. When applied to the mitochondria, the team found that it could be used to accurately measure its temperature while still inside the cell.
Mitochondria Are Physiologically Maintained At Close To 50 C, Dominique Chretien et al. biorxiv.org/content/early/2017/05/02/133223
444 notes
·
View notes

Conversation
Me: Let me go slip into somethimg more... comfortable.
Me: *comes back wearing a labcoat*
561 notes
·
View notes








Video
instagram
(via ScienceAlert (@sciencealert) • Instagram photos and videos)
Crabeater seal’s teeth are so incredible! They are like this so they can filter water and extract as much Antarctic krill as possible.
530 notes
·
View notes
Text
I think we should all take a moment to appreciate this gloriously well-preserved nodosaur that turned up in Alberta.

I mean, look at this thing!

This has got to be one of the most well-preserved dinosaurs I have ever laid eyes on.

Even the keratin sheaths around its osteoderm spikes were preserved, so it can be used to reliably reconstruct how ankylosaurids would have looked at in life!

And the best part is it was discovered completely by accident in a mining operation! Can you imagine being one of the miners who stumbled across this beautiful, beautiful thing?
National Geographic link
51K notes
·
View notes
Text
HEY WRITERS OF ALL KINDS AND AGES AND MAYBE EVEN DNDERS OR TABLETOP GAMERS ARE YOU READY FOR SOMETHING SUPER RAD? I HOPE SO ‘CAUSE
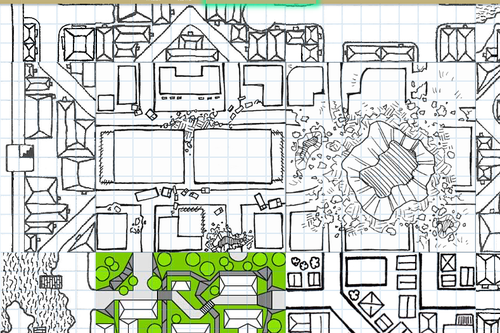
RANDOM
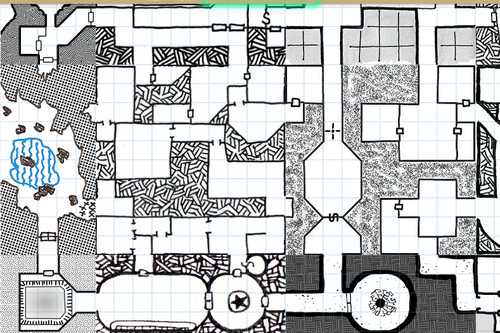
MAP
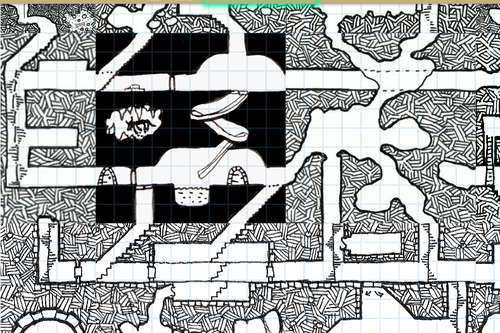
GENERATOR
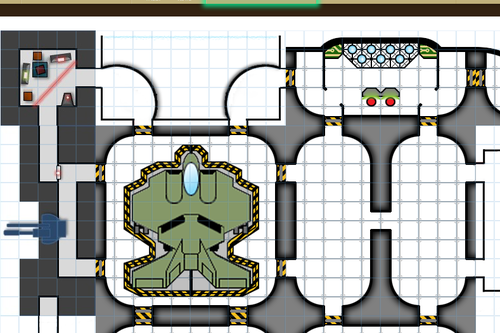
WITH

EDITING FEATURES AVAILABLE
IT DOESN’T REALLY DO LAND MASSES OR ANYTHING BUT IT SURE AS HELL WILL MAP THAT CITY/VILLAGE/SHIP/DUNGEON/WHATEVER THAT YOU’VE BEEN MEANING TO MAP OUT FOR YOU
SO FUCKING GO WILD
105K notes
·
View notes
Text
ALL MY WRITERS
I think the best piece of writing advice I ever got was from an author of locally popular novels that visited my school when I was in grade eight. He said that when you want to write a novel, or any kind of story, the typical system of “What is my story about? Who is it about? What will happen?” are pretty much the worst thing you can do.
Writing is far simpler than that.
His advice was to ask yourself three questions that I’ll never forget:
Who is the character?
What does the character want more than anything?
And how can I prevent them from getting it?
7K notes
·
View notes
Video
Without engineers, staying in shape would be a lot harder. #BeAnEngineer
12K notes
·
View notes
Text
site that you can type in the definition of a word and get the word
site for when you can only remember part of a word/its definition
site that gives you words that rhyme with a word
site that gives you synonyms and antonyms
1M notes
·
View notes
Photo

Got lots of comments on the berry I painted in this picture.
I hope this can help you learn a thing!
11K notes
·
View notes
Text
Temporarily Getting Rid of Characters
Do you ever have scenes with 6+ people and you can’t seem to juggle them all? Or do you ever want to get 2-3 characters in a scene together for some quality relationship development, but that means sending the other characters elsewhere?
I’ve compiled a list of “off-screen” things your character can be doing, for any time you need them to be temporarily somewhere else. With this list comes with a few caveats:
If you find yourself always sending the same character somewhere else to a point where that character never actually gets much page time, are you sure you actually need that character in your novel? Don’t use this an excuse to keep deadweight characters that are 100% unnecessary to your plot, but your heart can’t bear to cut. Either draw them back into the plot as a necessary element, or get rid of them.
This is only for temporarily getting rid of someone—meaning for a short amount of time, and infrequently. It starts to feel weird if your character always happens to be taking their nap when something happens, and if you’re not careful, it’ll come off as lazy writing. Use this kind of thing sparingly.
Sometimes, you’ll need to have a result to their temporary absence. For example, if you say they’re off spying on the bad guy, occasionally they need to come back with a tidbit of info. Not all of their missions will be a success, but again, it’ll start to feel fake if there’s no point to the spying.
And now to the list! There are two sections to it, depending on if your character to be doing something actively plot-related and useful while they’re away, or if you want them to stay uninvolved. Both have their uses depending on the situation.
Active:
Captured by the enemy
Fighting in a different area
Guarding a captured enemy
Protecting someone in potential danger
Researching the problem
Spying/recon
Training to fight
Watching someone suspicious
With another character (off-screen relationship building)
Passive:
At school/work
Doing a hobby
Hunting for food
Injured/sick/in the hospital
On vacation
Out of the loop (no one told them about the meeting, etc)
Sleeping
The passive list could be incredibly longer, but I tried to list enough for you to get the idea. Whatever you pick, make sure it makes sense in your plot and setting, and like I warned before, don’t use this as an excuse to hold onto deadweight characters.
Working this kind of downtime into your novel isn’t bad, since it can take up the necessary but boring actions that your story needs. Someone learning how to fight, for example, won’t learn everything in a week. By occasionally putting them “off-screen” to train, you’re further convincing us that he or she has trained enough to pull off a future scene where they hold their own in battle. It’ll also give a sense that other characters are working in the background and getting things done, even if your protagonist or narrator isn’t always there to see it, which is realistic, of course. It’s not like everyone else freezes in place the moment your protag isn’t around.
–E
11K notes
·
View notes
Photo

The Molecular and Cell Biology of Cancer - I
Cancer is a group of heterogeneous genetic diseases inherent in cells that proliferate in an unregulated manner. For a disease whose molecular characterisation began as recently as in the 70s, the span of these 4 decades have proven to be an aeon vis-a-vis breakthroughs in molecular oncology. Surprisingly, cancer holds an equally strong - almost ghoulish - fascination for the the general public, who probably think of it as the boogeyman of diseases; this is because, as real as the threat is, its scientific complexity continues to elude the masses. And yet it is everywhere, the poster child for hypochondria and morbidity alike: not only is it the go-to condition in popular literature and media, most of us can claim an acquaintance with someone affected. This apparent popularity may be justified, given that for roughly 14 million new cases diagnosed in 2012, 8.2 million existing patients died of cancer worldwide (1).
Through a series of articles, I will be writing about the science underlying carcinogenesis, about how genetic damage translates into molecular changes, which in turn shape the phenotypic hallmarks of the affected cells. Where applicable, this theoretical understanding will be complemented by the latest diagnostic and therapeutic applications.
So how does the process of tumour formation, called oncogenesis or tumourigenesis, begin? Essentially, the unrestricted growth and division that characterises cancerous cells can be attributed to endogenous (intrinsic) or exogenous (environmental) damage to our DNA, known as mutations. These mutations or genetic changes affect the production, structure and/or function of the molecules encoded by the affected genes. Therefore, oncogenesis has a genetic provenance, but at the cellular level, these changes manifest primarily as crosstalk between molecular components (usually *proteins*) of various signalling pathways within and between cells. *Figure 1 below comprehensively categorises the types of key proteins with a role to play in cancer cells*. The foremost goals of an altered signalling system are to overcome the Hayflick limit by repressing negative growth signals, and to favour survival over cell death (apoptosis); these two changes, along with other phenotypic alterations, are now known as the hallmarks of cancer (2). Collectively, these hallmarks confer a remarkable plasticity to cancer cells and will be discussed towards the end of this series.

Figure 1. The figure shows the chief protein classes involved in growth regulation, and thus cancer. Beginning from the membranous receptor proteins, signals are transduced to the nuclear machinery via downstream messenger proteins. The outcome of the nuclear gene expression further influences other cells by triggering their cell receptors, thus coming full circle.
The Genetics of Cancer
2015 began with an article in Science loosely citing “bad luck” as the major causative factor of cancer (3); the year ended with luck being trumped by exogenous (lifestyle and environmental) factors (4), more so than intrinsic damage. Regardless, the weakest links to take the first hit invariably remain the same: tumour suppressor genes (TSGs) and proto-oncogenes. TSGs are anti-oncogenes that act as a dual line of defence to mutations, in that they are recessive (5). In other words, both alleles need to undergo two successive mutations for oncogenesis to proceed unhindered. This principle is now known as Knudson’s Two-Hit hypothesis, and was first observed by cancer geneticist Alfred Knudson in the retinoblastoma gene (RB); briefly, children suffering from retinoblastoma (Rb) were found to harbour two non-functional RB alleles that caused either familial or sporadic forms of the tumour. Children who had already inherited one defective allele from a parent required only one somatic mutation to the second functional allele for familial retinoblastoma to manifest. Genomes with completely wild-type RB loci were found to require two somatic mutations to both RB alleles for the sporadic form of retinoblastoma to develop. Knudson thus concluded that oncogenesis required both alleles of certain genes, such as RB in this case, to be knocked out (Figure 2) (6). It must be noted that the lines between genetic dominance and recessivity are blurred in the RB gene: a heterozygous RB locus possessing a wild-type and a mutant allele (denoted as RB+/-) is recessive in that the cell remains phenotypically unaffected. But in the larger picture, this very heterozygosity acts dominantly, because it pre-disposes the carrier to lose the second unaffected allele, and thus develop retinoblastoma. Let’s address how the remaining wild-type RB allele is knocked out instead of a random target in a small population of cells.

Figure 2. The diagram illustrates the two modes in which mutations are acquired for retinoblastoma to develop.
In pre-cancerous cells, TSGs frequently undergo loss-of-function (LOF) in ways that circumvent the requisite second mutational event, because the chances of that happening are exceedingly rare, in the order of 10^-12 per base per generation (10^-6/gen. times 2, for two alleles). During mitotic cell division, the chromatids from a mother cell duplicate their genome, forming two diploid chromosomes. Before the mother can further divide into two daughter cells, a chromatid arm from each chromosome may cross over and exchange segments of DNA in a process termed mitotic recombination. If the regions containing the tumour suppressor (ex. RB) alleles recombine, there is a 50% chance that one of the two daughter cells will lose the functional allele as well (Figure 3). This condition is predictably called Loss of Heterozygosity (LOH) (7). The rate at which mitotic recombination results in LOH lies in the order of 10^-2 to 10^-4 per cell division (8), which is oftener than the aforesaid second-mutation probability of 10^-12 per generation. However, rates specific to different TSGs are not the same.

Figure 3. Mitotic recombination. The adjoining figure explains how mitotic recombination can result in crossover and the subsequent exchange of genetic information. To the far left are shown two homologous chromosomes that then duplicate their arms to form sister chromatids. The result of a crossover between 1 arm of each homolog is shown next, followed by the alternative fates of the daughter cells. The daughter cell to the far right ends up with homozygosity for the mutant alleles, shown in blue.
Even if the chromatid arms escape recombination, things can go awry in the next step, that is, the segregation of the 2 pairs of sister chromatids into two cells. Each cell must receive one haploid copy of each chromosome; however, non-disjunction results in one daughter cell receiving both sister chromatids in addition to one chromatid of the other chromosome, and the other daughter cell receiving only one chromatid. One of the three chromatids (now chromosomes!) is later degraded down that cell lineage to maintain diploidy, and if it is the normal chromosome that’s degraded, this confers a homozygosity at the mutant locus for that line (Figure 4) (7).

Figure 4. Non-disjunction of sister chromatids. In the figure, the sister chromatids (green) are shown to segregate into one daughter cell, and if the normal chromatid (red) is subsequently discarded, the cell line becomes homozygous for the mutant locus.
In accordance with the linked nature of the molecular chain of events, there’s always a ‘how’ or ‘why’ that proceeds a statement, and we might wonder why non-disjunction would occur in the first place. A not-too-onerous answer with molecular details will follow in a future article about cell cycles in cancerous cells, but to put things into perspective for the present, non-disjunction is a result of mis-segregation, i.e., an inability of the sister chromatids to separate, combined with the failure of the spindle assembly checkpoint to detect this error (7).This should demonstrate how cancer (or rather, onco-proteinic agents) is the perfect criminal, racing downstream via a parallel route and planning for all contingencies well in advance: not only do pre-cancerous cells possess a defective TSG allele, but they also facilitate the loss of the second allele by manipulating the expression of its upstream factors. In more optimistic terms, a single mutation or event is not enough to initiate and sustain cancer.
The third LOH mechanism is gene conversion, caused by the infidelity of the DNA polymerase (DNAP) that reads the DNA template during replication. Sometimes, the DNAP can promiscuously switch from its template strand to the strand of the homologous DNA sequence, thus reading the nucleotides of the wrong strand. If this misread region contains the mutated region, the new complementary strand will encode the inactive product (Figure 5) (7).

Figure 5. Gene conversion. The diagram illustrates the mechanism whereby DNA polymerase switches its choice of templates by temporarily shifting to the homologous chromosome (blue). Naturally, the unread segment in red will not be represented in the newly synthesised strand.
Epigenetics is the final major mechanism in TSG suppression; in an event that is as rare as a second somatic mutation, the proximal region of TSGs can be hyper-methylated by DNA methyltransferase enzymes at the promoter regions rich in CpG dinucleotides. These sites then recruit histone deacetylase enzymes complexed with methyl-binding domain proteins (HDAC/MBD). Histones are multimeric proteins that wrap around DNA and modulate its structural condensation; if the DNA is tightly packaged around these histones, the resulting molecule is inaccessible to other proteinic factors. If on the other hand, specific lysine residues on histone tails have been acetylated by the HDAC-antagonist histone acetylase (HAT), the positive lysines are neutralised by the acetyl group. This reverses the electrostatic attraction between the negative DNA backbone and the histones, and the DNA loosens into an open, transcribable form, known as euchromatin. Getting back to the point, HDACs strip the acetyl groups from lysine residues of the histones and revert them to their positively charged state. This disrupts the transcription factor (TF) binding sites formed by previously acetylated lysines, and TFs that promote expression of the downstream TSGs bind no more. Secondly, the now positive lysines are attracted to the negative DNA backbone and condense to form a closed chromatin structure, transforming from euchromatin to heterochromatin, which is not ideal for gene expression (Figure 6) (7). These mechanisms are referred to as transcriptional repression or gene silencing. The epigenetic ‘tags’, i.e. the methylated molecules, are exploited for diagnosis by methylation profiling of TSG promoters in tumour samples (9).

Figure 6. Euchromatin versus heterochromatin. The diagram compares the two alternative structures of DNA and its associated proteins. On the right, the histones wrapped round the DNA preceding the Transcriptional start site (TSS) are acetylated at lysine residues on their tail regions.
Arguably the most important tumour suppressor, called the guardian of the genome, is the protein p53 (Figure 7). Ironically, its gene TP53 was initially classified as a proto-oncogene by the company that p53 was found to keep; this 53 kDa protein co-precipitated in complex with the large T antigen of simian virus 40 (SV40 AgT) in cancerous hamster fibroblasts (10), besides interacting with the anti-apoptotic Adenovirus E1B oncoprotein and with the E6 oncoprotein of Papillomavirus (7). It is now known that these viral proteins interact with p53 to inactive its tumour suppression functions. Notwithstanding these viral oncoproteins, TP53 is also frequently mutated gene in many types of cancers that results in its loss of function - different p53 mutations are found in more than 50% of all human cancers, with more than 45,000 mutations already curated (11). Moreover, the TP53 gene locus has been traced to a frequently deleted region on chromosome 17 in colorectal cancer (11). Therefore it is now incontrovertibly known as a tumour suppressor.

Figure 7. p53 bound to DNA substrate. The tetrameric structure of p53 is illustrated (yellow and pink) engulfing the DNA, visualised in green-blue.
Both, pRb and p53 particularly crucial roles in cell cycle regulation, which explains why they are knocked out, inhibited, or targeted for degradation in cancer cell lines. Since this article has dealt with the mechanisms in which functional TSGs are lost, and taking into consideration that they are constantly involved in cancer cell metabolism, their manifold functions will be discussed over the course of future articles, as applicable.
REFERENCES
World Health Organisation [Internet] Cancer. [updated February 2015; cited 2016 January 14]. Available from: www.who.int/mediacentre/factsheets/fs297/en/
Hanahan D, Weinberg R. Hallmarks of Cancer: The Next Generation. Cell. 2011; 144[5]:646–74.
Tomasetti C, Vogelstein B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015; 347[6217]: 78-81.
Wu S, Powers S, Zhu W, Hannun Y. Substantial contribution of extrinsic risk factors to cancer development. Nature. nature; 2016;529[7584].
Pierce B. Scitable [Internet]. Proto-Oncogene [updated 2005; cited 2016 January 14]. Available from: http://www.nature.com/scitable/definition/tumor-suppressor-gene-184
Knudson AG. Two genetic hits (more or less) to cancer. Nature Reviews Cancer [Internet]. Nature Publishing Group; 2001;1(2):157–62. Available from: http://www.nature.com/nrc/journal/v1/n2/abs/nrc1101-157a.html
Weinberg RA. The Biology of Cancer. New York: Garland Science, Taylor & Francis Group; 2014.
California Institute of Technology [Internet] Mutations and Cancer lecture notes [cited 2016 January 17]. Accessed from: http://www.its.caltech.edu/~bi122/pdf/2015_Bi122_Lecture13.pdf
Lehmann DR, Gallie BL. NCBI [Internet]. GeneReviews Retinoblastoma [updated 2015 November 19; cited 2016 January 14]. Accessed from: http://www.ncbi.nlm.nih.gov/books/NBK1452/
Vogelstein B, Sur S, Prives C. Scitable [Internet]. p53 :The Most Frequently Altered Gene in Human Cancers; 2010. [cited 2016 January 15]. Available from: http://www.nature.com/scitable/topicpage/p53-the-most-frequently-altered-gene-in-14192717
Leroy B, Anderson M, Soussi T. TP53 mutations in human cancer: database reassessment and prospects for the next decade. Human mutation [Internet]. 2014; Available from: http://onlinelibrary.wiley.com/doi/10.1002/humu.22552/pdf
605 notes
·
View notes

