#desmielinización
Photo

La #esclerosismúltiple # caracteriza por focos diseminados de #desmielinización en el encéfalo y en la #médulaespinal. Sus síntomas más frecuentes incluyen alteraciones #oculomotoras, #parestesias, #debilidad, #espasticidad, #disfunciónurinaria y síntomas cognitivos leves. Lo típico es que los déficits #neurológicos sean múltiples, con remisiones y exacerbaciones que producen una #discapacidad paulatina. https://www.instagram.com/p/CkJ_91UDdDx/?igshid=NGJjMDIxMWI=
#esclerosismúltiple#desmielinización#médulaespinal#oculomotoras#parestesias#debilidad#espasticidad#disfunciónurinaria#neurológicos#discapacidad
1 note
·
View note
Text
Síndromes desmielinizantes agudos(SDA)
Las enfermedades desmielinizantes del sistema nervioso central (SNC) tienen un amplio espectro
de presentación y ocupan un capítulo muy importante dentro de la patología neurológica
en el adulto joven.
Las enfermedades desmielinizantes del sistema nervioso central (SNC) tienen un amplio espectrode presentación y ocupan un capítulo muy importante dentro de la patología neurológicaen el adulto joven. En los últimos años los episodios agudos de desmielinización delSNC con una topografía concreta han sido denominados Síndromes desmielinizantes aislados(SDA) haciendo referencia a un primer episodio…
View On WordPress
0 notes
Text
5. INMUNOSUPRESORES Y TOLÉROGENOS
En este espacio veremos los componentes de la respuesta inmune y los medicamentos que modulan la inmunidad a través de la inmunosupresión o tolerancia.
Pero, ¿Qué es el sistema inmune? ¿Qué es la inmunosupresión?
El sistema inmune se divide entre la inmunidad innata y la inmunidad adquirida.
Innata/natural: Es primitia y no requiere estimulación y es reactiva, en esta se encuentran granulocitos, monocitos, células NK, mastocitos y básofilos.
Adquirida/aprendida/adaptativa: Depende de la exposición antigénica o sensibilización del antígeno y tiene alta afinidad, aquí encontramos los linfocitos B y T.
La inmunosupresión son medicamentos que se utilizan para amortiguar la respuesta inmune en el trasplante de órganos y enfermedad autoinmune.
Los tolerógenos son los que se utilizan para inducir tolerancia y tienen incapacidad para producir una respuesta específica.
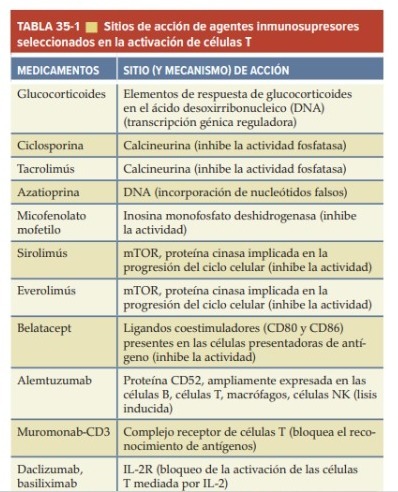
Para trasplante los medicamentos inmunosupresores son:
Glucocorticoides
Inhibidores de calcineurina
Agentes antiproliferativos/antimetabólicos
Biológicos (anticuerpos)
Sitios de acción:
Glucocorticoides:
La prednisona, prednisolona y otros glucocorticoides se
usan solos y en combinación con otros agentes inmunosupresores para el tratamiento del rechazo de trasplantes y enfermedades autoinmunes.
Tienen amplios efectos antiinflamatorios en varios
componentes de la inmunidad celular, pero relativamente poco efecto
sobre la inmunidad humoral. Los glucocorticoides se unen a receptores dentro de las células y regulan la transcripción de otros muchos genes.
Inhibidores de la calcineurina:
los cuales se orientan a las vías de señalización intracelular inducida como consecuencia de la activación de TCR. La ciclosporina y el
tacrolimús se unen a una inmunofilina.
Tacrolimús:
Debido a la eficacia percibida un poco mayor y a la facilidad
para vigilar las concentraciones sanguíneas, el tacrolimús se ha convertido en el inhibidor de la calcineurina preferido en la mayoría de los centros de trasplante.
Inhibe la activación de las células T inhibiendo la calcineurina. El tacrolimús se une a una proteína intracelular.
Ciclosporina:
Polipéptido cíclico de 11 aminoácidos, producida por el hongo Beauveria nivea, que inhibe la actividad de la calcineurina. Se une a la calcineurina, inhibiendo la desfosforilación estimulada por el Ca2+}
Activación de las células T y sitios de acción de agentes inmunosupresores.
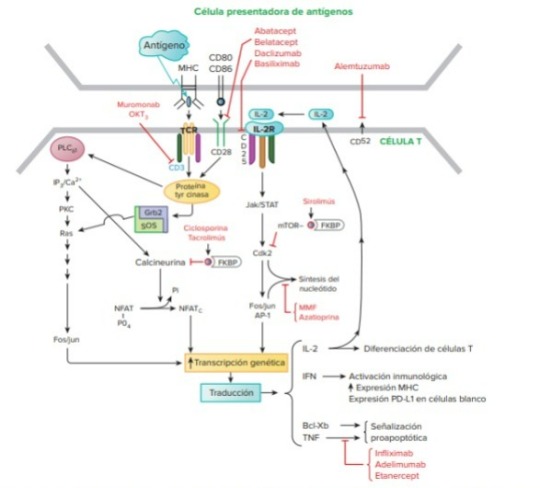
Medicamentos antiprofeliferativos y antimetabólicos:
Sirolimús
Everolimús

Azatioprina

Micofenoldo mofetilo

Agentes antiproliferativos y citotóxicos:
Fingolimod
Globlina antitimocito
Anticuerpos monoclonales Inmunoterapia y la naturaleza de la coestimulación y la inhibición:
Anticuerpos monoclonales anti-CD3
Anticuerpo monoclonal anti-CD52 (alemtuzumab)
Anticuerpos antirreceptores de IL-2 (anti-CD25)
elatacept, una proteína de fusión
youtube
Tolerancia:
La tolerancia, representaría una cura verdadera para los trastornos discutidos previamente en esta sección sin los efectos de
varias terapias inmunosupresoras.
Inmunoterapia para la esclerosis múltiple:
youtube
La esclerosis múltiple es una enfermedad inflamatoria desmielinizante
de la materia blanca del CNS mediada genéticamente, caracterizada por infiltración de células mononucleares en la materia blanca con la relativa desmielinización y la pérdida axonal.
En la farmacoterapia:
Las terapias específicas están dirigidas a resolver ataques agudos, reducir las recurrencias y las exacerbaciones, y retrasar la progresión de la discapacidad. Las exacerbaciones de la MS se tratan con 3 a 5 días de 1000 mg de metilprednisolona por vía intravenosa, ya que la prednisona oral sola es un tratamiento ineficaz que aumenta el riesgo de nuevos ataques.
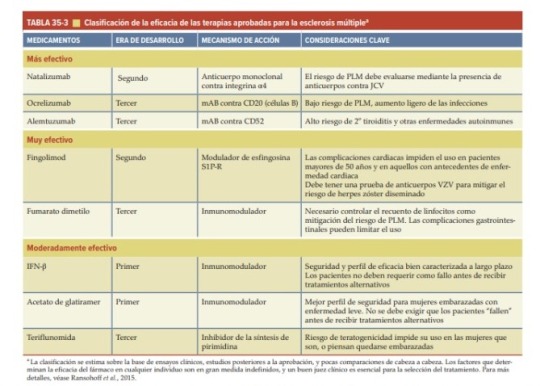
Datos para la eficacia de las terapias:
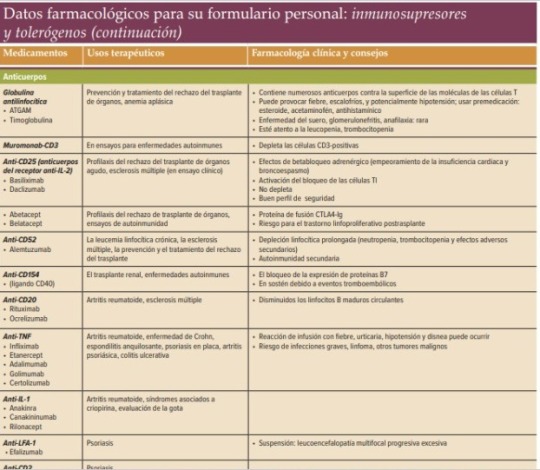
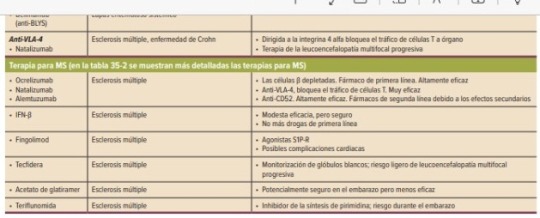
Datos completos de fármacos para la inmusupresión y tolerógenos.


43 notes
·
View notes
Photo

Dɪғᴛᴇʀɪᴀ
A fines del siglo XIX miles de personas morían anualmente a causa de una de las grandes epidemias del momento, el tétanos y la difteria, gracias a esto personas como Robert Koch, Louis Pasteur y Emil Von Behring dedicaron parte de su vida a la búsqueda de su solución, dando este último con ella, la vacuna de para el gran problema que había estado afectando mayormente a la población infantil; de esta manera se dio también inicio al desarrollo de la Inmunología.
La difteria es una infección causada por la bacteria Corynebacterium Diphtheriae. Sus signos y síntomas, que suelen manifestarse entre 2 y 5 días después de la exposición, pueden ser desde leves hasta graves. A menudo los síntomas se presentan de modo gradual, empezando por dolor de garganta y fiebre.
la difteria afecta más comúnmente a la mucosa respiratoria, provocando apariencia hiperemia local del edema de la mucosa, seguido por necrosis y la inflamación aguda.
El organismo se multiplica la puerta, mientras que la liberación de la toxina diftérica. La toxina actúa tanto a nivel local como sistémica. Esto ayudará a producir pseudomembrane local. Sistémica después de la absorción en la sangre hará que la miocarditis, neuritis y nocroza en diversos órganos (riñón, hígado, suprarrenales). Si la acción fue destructiva, la difteria puede dañar de forma permanente el miocardio; También el sistema nervioso causará la desmielinización de las estructuras nerviosas periféricas.
La toxina de la difteria se compone de dos fragmentos: el fragmento A y fragmento B, el último es responsable de los fenómenos de daño a los nervios local y el fragmento A se produce. Una sola molécula de fragmento penetrado en el citoplasma de la célula puede matar.
En los casos de gravedad, la bacteria genera un producto tóxico (toxina) que da lugar a una espesa placa gris o blanca en el fondo de la garganta, placa que a veces, al bloquear las vías respiratorias, dificulta la respiración o la deglución y también puede provocar una tos seca.La hipertrofia de los ganglios linfáticos puede causar la hinchazón de una parte del cuello.
A veces la toxina pasa al torrente sanguíneo y causa complicaciones como inflamación y lesión del miocardio, inflamación de los nervios, problemas renales o trastornos hemorrágicos por caída del nivel de plaquetas. Las lesiones del miocardio pueden provocar alteraciones del ritmo cardíaco e inflamaciones de los nervios que a veces desembocan en parálisis.
En la actualidad, la difteria es rara en los países desarrollados porque es habitual la inmunización en la niñez. Sin embargo, después del desmembramiento de la ex Unión Soviética, las tasas de vacunación en los países que la formaban disminuyeron, y posteriormente aumentaron mucho los casos de difteria. La susceptibilidad también ha aumentado porque las tasas de inmunización de refuerzo están disminuyendo en los adultos.
Bush L et al . (2017). Difteria. Agosto 21 del 2019, de Manual MSD Sitio web: https://www.msdmanuals.com/es/professional/enfermedades-infecciosas/bacilos-grampositivos/difteria#v1005971_es
OMS. (2017). Difteria . Agosto 26 del 2019, de OMS Sitio web: https://www.who.int/features/qa/diphtheria/es/
2 notes
·
View notes
Text
Dietas y salud ósea: ¿comer carne roja puede prevenir la esclerosis múltiple?

Esclerosis: El vínculo entre ciertas dietas y la esclerosis múltiple (EM) es un área de gran interés entre los miembros de la comunidad de EM.
Esclerosis: Si bien ya hay estudios establecidos que apuntan a los efectos beneficiosos de ciertos alimentos y componentes dietéticos como los ácidos grasos omega-3 , no hay suficiente investigación para llegar a un consenso sólido sobre si una dieta en particular es beneficiosa para las personas con EM.

Ahora, la evidencia reciente sugiere que una dieta mediterránea podría ser la clave para el tratamiento dietético de la EM.
Un estudio publicado en el Journal of Nutrition descubrió que una dieta mediterránea que incluye carne roja sin procesar puede reducir el riesgo de desarrollar desmielinización del sistema nervioso central (SNC), que es un precursor común de la EM.
Esclerosis: La dieta mediterránea y la carne roja para la prevención de la EM
Según la National Multiple Sclerosis Society, aproximadamente un millón de adultos mayores de 18 años en los EE. UU. se ven afectados por la EM.
Los expertos afirman que los factores de riesgo para desarrollar EM también incluyen factores ambientales como la baja exposición al sol, baja vitamina D y una dieta pobre.
Sin embargo, la evidencia que vincula la dieta con el riesgo de EM no es concluyente.
Esclerosis: “Investigaciones anteriores sugieren que una dieta mediterránea puede ayudar a reducir el riesgo de ciertos problemas de salud, como enfermedades cardiovasculares, diabetes, cáncer, enfermedad de Alzheimer y demencia, y mejorar la esperanza de vida en general.

Sin embargo, no hay pruebas concluyentes que sugieran que una dieta mediterránea también reduzca el riesgo de desarrollar EM ", dijo la autora principal, Lucinda Black, de la Facultad de Salud Pública de la Universidad de Curtin .
Debido a esto, los investigadores de Australia intentaron examinar las asociaciones entre una dieta mediterránea y el riesgo de un primer diagnóstico clínico de desmielinización del SNC.
Para hacerlo, el equipo de investigación reunió y analizó datos del estudio Ausimmune, un estudio multicéntrico de casos y controles que examina los factores de riesgo ambientales para la desmielinización del SNC.
Los datos de este estudio incluyeron 282 casos de personas con EM junto con 558 sujetos de control sanos.
Black y su equipo utilizaron el puntaje alternativo de dieta mediterránea (aMED) para medir la adherencia de los participantes a las dietas, siendo 9 la adherencia más alta y 0 la más baja.
El equipo también desarrolló una versión modificada de este puntaje llamada aMED-Red, con un punto asignado a los participantes que comieron aproximadamente una porción de 65 gramos de carne roja sin procesar.
Luego, los investigadores dividieron a todos los participantes en cuatro categorías: Categoría 1 (0-2 puntos), categoría 2 (3-4 puntos), categoría 3 (5 puntos), categoría 4 (6-9 puntos).
Los resultados revelan que, si bien no existe una asociación estadísticamente significativa entre aMED y el riesgo de desmielinización del SNC, vieron que las personas en los niveles superiores de los puntajes de aMED-Red mostraron un riesgo significativamente menor de desmielinización del SNC correspondiente a un 37 por ciento, 52 por ciento, y 42 por ciento menos riesgo de desmielinización del SNC en las categorías 2, 3 y 4, respectivamente.
Esclerosis: "Nuestra investigación encontró que consumir una porción diaria (65 g) de carne roja sin procesar como parte de una dieta mediterránea saludable puede ser beneficioso para aquellos con alto riesgo de desarrollar EM", dijo Black.

"No está claro por qué consumir carne roja combinada con una dieta saludable puede reducir el riesgo de EM, pero la carne roja contiene macro y micronutrientes importantes, como proteínas, hierro, zinc, selenio, potasio, vitamina D y una gama de vitaminas B, muchos de los cuales son importantes para una función neurológica saludable".
Según la coautora Robyn Lucas de la Universidad Nacional de Australia en Canberra, esta investigación destacó la necesidad de educar a las personas que corren un mayor riesgo de desarrollar EM sobre el impacto de su dieta en su salud general.
Tags
esclerosis multiple tratamiento, síntomas de esclerosis múltiple.
esclerosis multiple sintomas, esclerosis sintoma, esclerosis degenerativa.
esclerosis multiple sintomas tempranos, esclerosis multiple pdf.
Read the full article
0 notes
Photo

Lorenzo´s oil (1992)
Director: George Miller.
Género: Drama.
Duración: 135 min.
Sinopsis:
Hasta aproximadamente la edad de 7, Lorenzo Odone era un niño normal. Después de eso, cosas extrañas comenzaron a sucederle: tendría apagones, lapsos de memoria y otros extraños fenómenos mentales. Eventualmente se le diagnostica que sufre de ALD: un trastorno cerebral degenerativo incurable extremadamente raro. Frustrados por las fallas de los médicos y de la medicina en esta área, los Odones comienzan a educarse en la esperanza de descubrir algo que pueda detener el progreso de la enfermedad.
Esta historia comienza en África con una despedida, una familia de procedencia Italiana que vive en África debido al trabajo del padre se muda ahora a Estados Unidos por esto mismo, el hijo Lorenzo Odone se despide de su amigo Africano Omuri a través de cometas con dibujos de él y de sus padres. Al llegar a Estados Unidos es cuando los síntomas de Lorenzo comienzan a manifestarse, sus profesoras y los miembros de la comitiva de la escuela advierten a su madre Michaela y a su padre Augusto sobre esto, pensando que podía ser consecuencia de problemas familiares lo cual los padres negaron pero no fue hasta después de un episodio de Lorenzo en su casa que sus padres se dieron cuenta de que estaba teniendo episodios en los que actuaba con muy poco control de sus facultades y tenía comportamientos destructivos, buscaron diagnósticos y pensaron que era trastorno de déficit de atención, pero para los Odone era muy extraño que un niño tan inteligente como su hijo y que incluso sabía 3 idiomas pudiera sufrir de esto, a pesar de esto tuvieron que darle educación especial.Después de un tiempo el niño presento más síntomas, se cayó de su bicicleta en navidad y se lastimo, también de una silla y demás, le hicieron análisis después de internarlo en varias ocasiones en la clínica y por fin obtuvieron una respuesta, Lorenzo tenia ALD (Adenoleucodistrofia), una enfermedad hereditaria transmitida por el cromosoma x incluida en el grupo de las leucodistrofias. Produce una desmielinización intensa y la muerte prematura en niños, y la adrenomieloneuropatía se asocia a una neuropatía mixta, motora y sensorial, con paraplejía espástica en los adultos. Ambos procesos cursan con niveles circulantes elevados de ácidos grasos de cadenas muy largas que provocan insuficiencia suprarrenal. Los médicos dicen que la enfermedad es contundente e intratable, pero sus padres no se dan por vencidos.
Los Odone dedican sus días a estudiar e investigar la ALD y sus tratamientos para encontrar una manera de ayudar a Lorenzo, acuden a grupos de ayuda para familias de pacientes con ALD, también visitan numerosos médicos para consultar segundas opiniones, pero no obtienen resultados. Se embarcan en un estudio de seis meses que consiste en el retrasar el desarrollo de la enfermedad cambiando la dieta de los niños para disminuir los ácidos grasos de cadena larga C24 y C26 pero al pasar dos veces ven que no hay resultados. Empíricamente descubren en estudios de animales que una forma de reducir los niveles de C24 y C26 es produciendo otro acido que sea menos dañino, este es el ácido oléico monoinsaturado C18, componente habitual del aceite de oliva así que sus padres proponen la idea a los médicos en la primera conferencia de ALD, pero estos no la aprueban ya que puede ser peligrosa y no están confirmados los resultados, a pesar de esto los Odone consiguen el aceite y comienzan a tratar a Lorenzo con él, esto ayuda mucho ya que disminuye sus niveles de C24 y C26 en un 50%. Pero el avance se estanca y Lorenzo se sigue deteriorando, cada vez tiene más dificultades, ya no se mueve ni habla, le cuesta tragar incluso su saliva y además de esto, mantener su calidad de vida es bastante trabajo para sus padres y su tía, además de las múltiples enfermeras que por lo general no se ajustan a lo que busca la Sra. Odone.
Después de un tiempo el Sr. Odone siguen buscando tratamientos y es cuando descubre que una misma enzima se encarga de sintetizar los ácidos grasos monosaturados y alargar los saturados. Se plantean el usar la inhibición enzimática competitiva para bloquear el alargamiento los ácidos grasos saturados por encima de C22 y C24, añadiendo ácido erúcico. Esta idea tampoco es muy bien recibida por la comunidad médica, los cuales deciden que los factores en contra son más que los que tiene a favor. Ellos siguen adelante y buscan quien los ayude a aislar este aceite, al lograrlo con un químico extranjero comienzan a tratar a Lorenzo y logran que sus niveles de C24 y C26 sean normales, es un momento muy importante para sus padres, su tía y su amigo Omuri que vino desde África a cuidarlo.
Al final de la película se muestra como Lorenzo desarrolla la capacidad de comunicarse al cerrar los ojos (A los 12 años) y se menciona que continua progresando, así como que el aceite ayudo a cientos de niños más, (como al segundo hijo de la amiga de su madre que también estaba enfermo) y también que su padre Augusto continua investigando para realizar operaciones que permitan restablecer la mielina a nivel cerebral con trasplantes de células neuronales que hasta el momento solo se habían planteado en animales.
Considero que esta es una historia de la dedicación, el amor y la devoción que pueden tener unos padres con su hijo, ellos superan una tras otra dificultad y nunca pierden la esperanza de que las cosas mejoren para ayudar a sus hijos y a otros niños, en esta película vemos a muchos médicos que se involucran en el caso y que a pesar de querer ayudar y hacer lo mejor para ayudar a Lorenzo no pueden hacerlo debido a que existen lineamientos y normas que deben ser seguidas, pruebas y procedimientos que deben ser realizados antes de implementar nuevos tratamientos sin saber sus repercusiones, por lo cual no pueden cooperar mucho con la investigación de los Odone ya que el método científico no puede ser alterado.
1 note
·
View note
Text
Marcador genético para el riesgo de drogas en la esclerosis múltiple ofrece camino hacia la medicina de precisión
Marcador genético para el riesgo de drogas en la esclerosis múltiple ofrece camino hacia la medicina de precisión

Desmielinización por MS. El tejido de color CD68 muestra varios macrófagos en el área de la lesión. Escala original 1: 100. Crédito: Marvin 101 / Wikipedia
Un equipo de investigadores ha descubierto una variante genética específica asociada con una reacción adversa al medicamento que resulta en daño hepático en personas con esclerosis múltiple (EM). Es…
View On WordPress
0 notes
Text
Esclerosis múltiple
Esclerosis múltiple
La esclerosis múltiple (EM) es una enfermedad del sistema nervioso. En la EM, hay defectos en la comunicación entre el cerebro y otras partes del cuerpo. Los efectos de la EM pueden variar desde relativamente leves en la mayoría de los casos hasta algo incapacitantes o devastadores. Los síntomas pueden ocurrir de manera aleatoria (en brotes) y luego desaparecer. En el peor de los casos, una persona con EM puede ser incapaz de caminar, hablar o escribir.
Durante un brote de EM, la inflamación ocurre en parches (llamados placas) en cualquier área del sistema nervioso. Esta inflamación causa daño a la vaina de mielina. La vaina de mielina es una cubierta de grasa que protege las fibras nerviosas en el cerebro y la médula espinal. La mielina permite la transmisión suave y de alta velocidad de señales nerviosas entre el cerebro, la médula espinal y el resto del cuerpo. Cuando la mielina se daña, puede bloquear o ralentizar las señales nerviosas, lo que resulta en una reducción o pérdida de la función.
Signos y síntomas
Los síntomas de la EM incluyen:
Debilidad en brazos y piernas
Problemas con el equilibrio y la coordinación
Cansancio
Hormigueo o entumecimiento en cualquier área
Mareos
Visión borrosa
Dolor
Sensibilidad al calor
Pérdida del control de la vejiga
estreñimiento
Pérdida de memoria
Dificultad para resolver problemas
Alteraciones del estado de ánimo
Temblores
Espasmos musculares
Problemas del habla
Dificultad para deglutir
¿Qué Causa la esclerosis múltiple?
Se desconoce la causa exacta de la EM. Los científicos piensan que la enfermedad es una condición autoinmune influenciada por factores genéticos y ambientales. Otras teorías involucran factores bacterianos o virales.
¿Quién tiene más riesgo?
Las personas con las siguientes afecciones o características tienen mayor riesgo de desarrollar EM:
Familiares de primer grado con EM
Edad entre 20 a 40 años
Vivir en las latitudes del norte durante los primeros 15 años de vida
Ascendencia norteeuropea, norteamericana o escandinava
Genes de respuesta inmunitaria
Género femenino
Fumar cigarrillos
Deficiencia de vitamina D
Qué esperar en el consultorio de su médico
Si tiene síntomas asociados con la EM, debe consultar a su proveedor de atención médica. Su proveedor lo hará:
Pedir historia detallada
Verificar si hay problemas neurológicos
Compruebe su visión
El proveedor puede ordenar los siguientes exámenes:
Prueba de estudio de la función nerviosa (prueba del potencial evocado)
Exámenes de laboratorio, como un examen del líquido cefalorraquídeo (LCR) por punción lumbar
Procedimientos de diagnóstico por imágenes, como la resonancia magnética (IRM)
Exámenes de sangre para descartar otras afecciones
Opciones de tratamiento
Plan de tratamiento
No existe una cura conocida para la EM en este momento. El objetivo principal del tratamiento es reducir los síntomas de control y mejorar la calidad de vida.
Terapias con medicamentos
El médico puede prescribir los siguientes medicamentos o una combinación de ellos:
Esteroides para reducir la gravedad de los ataques
Interferón beta para disminuir la destrucción de la mielina, reducir la frecuencia y severidad de los ataques y la progresión lenta de la enfermedad.
La inmunoterapia y las citoquinas son terapias experimentales que pueden alterar el curso de la enfermedad.
Terapias complementarias y alternativas
Aunque ninguna terapia complementaria o alternativa puede curar, tratar o prevenir la EM, algunas estrategias pueden mejorar los síntomas de la EM. Sin embargo, algunas terapias CAM pueden interferir con los tratamientos convencionales. Informe a todos sus proveedores acerca de cualquier terapia CAM que esté considerando.
Nutrición y Suplementos: Estos consejos nutricionales pueden ayudar a reducir los síntomas:
Consuma una dieta baja en grasas saturadas.
Consuma una dieta rica en fibra, particularmente de granos enteros, frutas y verduras. Las dietas altas en fibra son importantes para prevenir el estreñimiento.
Reduzca el consumo de carne roja e incluya pescado en su dieta.
Evite el alcohol.
Deje de fumar.
Se recomienda una dieta multivitamínica.
Los estudios epidemiológicos han encontrado una disminución de la incidencia de la EM en poblaciones que tenían un bajo consumo de grasas animales con un alto consumo de pescado de agua fría.
Se recomienda hacer ejercicio de leve a moderado, por lo menos 30 minutos diarios durante aproximadamente 3 veces a la semana.
El Protocolo Wahls. Una intervención dietética para el tratamiento de la EM y otras enfermedades autoinmunes. Fue creado por Terry Wahls, MD, quien a su vez tenía EM y fue capaz de resolver la gran mayoría de sus síntomas utilizando un enfoque dietético especializado. Ella lleva a cabo estudios de investigación sobre este enfoque y está ganando popularidad. Hable de esta opción con su proveedor.
Usted puede tratar las deficiencias nutricionales con lo siguiente:
Ácidos grasos omega-3. Los aceites de pescado tienen un alto contenido de ácidos grasos omega-3. Los peces de agua fría (por ejemplo, arenque, salmón o caballa) son ricos en ácidos grasos omega-3. Éstos tienen efectos antiinflamatorios que benefician a Maty en la EM.
aceite de onagra. Es rico en ácidos grasos omega-6 y es comúnmente utilizado por pacientes con EM. Sin embargo, los estudios no lograron probar ningún beneficio del aceite de onagra para la EM. El aceite de onagra puede interferir con varios medicamentos.
Multivitamínico/multimineral. En general, se recomienda que las personas con EM contengan las vitaminas antioxidantes A, C, D, E, las vitaminas B y los oligoelementos, como el Magnesio, el calcio, el zinc y el Selenio.
Vitamina D. Los científicos han identificado la deficiencia de vitamina D como un posible factor que contribuye al desarrollo de la EM. La exposición al sol y las fuentes dietéticas de vitamina D durante la infancia y la adolescencia se han asociado con un menor riesgo de desarrollar EM. La deficiencia de vitamina D puede estar asociada con un empeoramiento de los síntomas en las primeras etapas de la EM.
coenzima Q10. Puede tener efectos antioxidantes y antiinflamatorios. Sin embargo, ningún estudio ha encontrado que la coenzima Q sea efectiva en la EM. La coenzima Q10 puede interactuar con ciertos medicamentos anticoagulantes, como la wafarina (Coumadin) y otros.
N-acetil Cisteína. Tiene efectos antioxidantes y es utilizado por algunos pacientes con EM. No existen estudios que demuestren los efectos de la N-acetil cisteína en el tratamiento de la EM. La N-acetilcisteína puede interactuar con la nitroglicerina.
Acetil-L-Carnitina. También puede tener efectos antioxidantes. Algunos estudios apoyan el uso de acetil-L-carnitina en personas con EM. Sin embargo, algunos investigadores han sospechado que la acetil-L-carnitina no es segura para las personas con antecedentes de trastornos convulsivos. Hable con su médico antes de tomar acetil-L-carnitina.
Suplemento probiótico (que contiene Lactobacillus acidophilus). Puede ayudar en el mantenimiento de la salud gastrointestinal e inmunológica. Varios estudios sugieren que los probióticos pueden reducir la inflamación en la EM y, por lo tanto, podrían ser beneficiosos. Sin embargo, los probióticos no son apropiados para individuos que están severamente inmunosuprimidos o que están tomando medicamentos inmunosupresores.
Melatonina. Tiene efectos antioxidantes y antiinflamatorios. También se demostró que actúa como un regulador inmunológico y algunos estudios demostraron que puede mejorar el curso de la EM al inhibir la desmielinización y aumentar la remielinización. Se necesitan más estudios. La melatonina puede interactuar con muchos medicamentos, incluyendo sedantes, antidepresivos, medicamentos hormonales, incluyendo los anticonceptivos, y otros.
Las hierbas no puede curar la esclerosis múltiple ni sus complicaciones. Sin embargo, algunas hierbas pueden ayudar con ciertos síntomas y se pueden usar además de la terapia convencional. Las hierbas pueden tener efectos secundarios e interactuar con otras hierbas, suplementos o medicamentos. Por estas razones, usted debe tomar las hierbas con cuidado, bajo la supervisión de un médico. Aunque con la excepción del cannabis, los estudios clínicos no apoyan el uso de hierba para el tratamiento de la EM, algunas de las hierbas utilizadas por las personas con EM incluyen:
Té verde (Camellia sinensis). Puede tener efectos antioxidantes e inmunológicos. Algunos estudios probaron los efectos de los compuestos del té verde llamados catequinas sobre la debilidad muscular en la EM. Se necesita más investigación. El té verde consumido como bebida es generalmente seguro, sin embargo, los extractos concentrados de té verde pueden tener efectos tóxicos en el hígado.
Rhodiola (Rhodiola rosea). Puede tener efectos antioxidantes e inmunológicos. Algunas personas con EM utilizan Rhodiola para mejorar los síntomas de la fatiga y la intolerancia al ejercicio; sin embargo, no existen estudios que la apoyen. Hable con su proveedor.
Semillas de cardo mariano (Silybum marianum). A veces se utiliza para mejorar los síntomas de la enfermedad hepática. Algunos estudios sugieren que la silimarina, el principal componente de las semillas de cardo lechoso, tiene efectos antiinflamatorios. El cardo mariano puede causar una reacción alérgica en personas sensibles a la ambrosía. Debido a que el cardo lechoso funciona en el hígado, puede interferir potencialmente con una variedad de medicamentos. Hable con su médico.
bromelina (Ananus comosus). A veces se utiliza para el dolor y la inflamación. La bromelina es una mezcla de enzimas digestivas de proteínas que se encuentran en el tallo de la piña. En estudios de laboratorio, la bromelaína tuvo efectos antiinflamatorios. La bromelaína puede aumentar los efectos adelgazadores de la sangre de ciertos medicamentos, como la warfarina (Coumadin) y la aspirina, y puede interferir con ciertos medicamentos, incluyendo algunos antibióticos.
Ginkgo biloba. Se ha utilizado en la medicina tradicional china para pacientes con deterioro cognitivo y Demencia. Estudios clínicos encontraron que el Ginkgo biloba puede reducir la fatiga en personas con EM. No se han confirmado otros beneficios para la salud. Los efectos secundarios incluyen dolores de cabeza y reacciones alérgicas. El ginkgo biloba interactúa con algunos medicamentos, incluyendo los anticoagulantes. Algunos estudios de laboratorio encontraron que los animales expuestos al Ginkgo biloba tenían un mayor riesgo de padecer algunos tipos de cáncer. Comer semillas crudas o tostadas de Gingko biloba puede tener efectos secundarios peligrosos. Asegúrese de consultar con su proveedor antes de tomar Ginkgo biloba.
ginseng asiático (Panax ginseng). Los compuestos que se encuentran en el ginseng pueden tener efectos en el sistema inmunológico. Algunos estudios encontraron que el ginseng puede ayudar a tratar la fatiga asociada con la EM, pero se necesitan más resultados clínicos. El ginseng puede afectar la presión arterial y el azúcar en la sangre y podría interactuar con medicamentos como los anticoagulantes. Hable con su proveedor antes de tomar ginseng.
Extracto de semilla de uva (Vitis vinifera). Contiene sustancias antioxidantes y es utilizado por algunas personas con EM. No existen estudios clínicos que apoyen el extracto de semilla de uva para el tratamiento de los síntomas de la EM. El extracto de semilla de uva es generalmente seguro, aunque su seguridad no está clara en personas con trastornos hemorrágicos o que toman anticoagulantes. Consulte con su proveedor.
Cannabis sativa. Ha sido demostrado como una terapia efectiva para las personas con EM en múltiples estudios clínicos. Los cannabinoides (ya sea como extracto oral, aerosol o tetrahidrocannabinol sintético) se encuentran actualmente entre los tratamientos clínicamente recomendados para el tratamiento del dolor y la espasticidad en la EM. Las recomendaciones no incluyen el cannabis fumado (marihuana), sobre el cual los resultados de los estudios no son claros.
Los efectos secundarios son generalmente leves para el uso a corto plazo. A largo plazo, debido a sus propiedades psicoactivas, la seguridad del uso de los cannabinoides es incierta. En los Estados Unidos, el estatus legal de las preparaciones específicas de cannabis medicinal varía entre los diferentes estados, y la FDA no ha aprobado ningún producto que contenga o se derive de la marihuana botánica para ninguna indicación.
Homeopatía Ningún estudio clínico encontró que los remedios homeopáticos fueran efectivos en el tratamiento de la EM. Sin embargo, algunas personas con EM utilizan un enfoque homeopático junto con el tratamiento convencional para algunos de sus síntomas. Los remedios incluyen:
Carboneum sulphuratum
Causticum
Lathyrus sativus
fósforo
Medicina física Terapia de ejercicio. El ejercicio físico mejora varios síntomas de la EM. El ejercicio puede disminuir las complicaciones de la debilidad muscular y la espasticidad. El ejercicio también mejora la salud general y el bienestar. Los ejercicios acuáticos pueden ser de especial utilidad para las personas con EM. El ejercicio es generalmente seguro y bien tolerado.
Acupuntura. La acupuntura puede aliviar los síntomas de la EM. Los estudios pequeños mostraron beneficios de la acupuntura en pacientes con EM, pero se necesitan estudios clínicos más grandes y de mayor calidad para probar su eficacia y seguridad.
Masaje. El masaje podría ser útil para mantener la flexibilidad y reducir la espasticidad, así como para mejorar la sensación general de bienestar.
Yoga. Los estudios sobre el efecto del yoga en los síntomas de la EM fueron inconsistentes. Algunos estudios observaron efectos beneficiosos sobre la movilidad, la fatiga y la calidad de vida. Otros estudios no encontraron ningún beneficio adicional en comparación con otras formas de ejercicio. El yoga se considera generalmente seguro.
Pronóstico / Posibles complicaciones
Al principio de la EM, alrededor del 85% de las personas experimentan ataques separados por períodos de remisión, durante los cuales los síntomas disminuyen. Aproximadamente la mitad de estas personas tienen un empeoramiento crónico y progresivo después de 10 a 15 años. Alrededor del 15% de las personas experimentan un empeoramiento progresivo crónico desde el inicio.
La mayoría de las personas con EM viven durante 30 años o más con la enfermedad. La mayoría de las personas con EM son activas y funcionan en el trabajo con poca discapacidad. El grado de discapacidad e incomodidad depende de:
La frecuencia y la gravedad de los ataques
La parte del sistema nervioso que es afectada por cada ataque
La mayoría de las personas regresan a sus funciones normales o casi normales entre los ataques. Con el tiempo, hay una mayor pérdida de función con menos mejoría entre los ataques.
Las disfunciones vesicales, intestinales y sexuales son comunes entre esta población. Otras complicaciones pueden incluir
Dificultad para deglutir
Úlceras por presión
osteoporosis
Dificultad para pensar
Depresión
Infecciones del tracto urinario
Efectos secundarios de los medicamentos utilizados para tratar la afección
Seguimiento
Las personas con EM necesitarán un seguimiento de por vida, especialmente durante los brotes.
from WordPress https://homeopatiaynaturopatia.com/esclerosis-multiple/?utm_source=rss&utm_medium=rss&utm_campaign=esclerosis-multiple
0 notes
Text
Síndrome de Guillain-Barré: síntomas, causas y tratamiento
**El síndrome de Guillain-Barré es una enfermedad rara que destruye la mielina de los nervios periféricos** del organismo y provoca alteraciones musculares y sensitivas, generando en la persona que la sufre una gran discapacidad funcional. Es un trastorno grave que debe abordarse con urgencia, ya que puede derivar en complicaciones respiratorias que pueden poner en riesgo la vida del paciente. En este artículo te explicamos en qué consiste esta enfermedad neurológica, cuáles son sus causas, los signos y síntomas, cómo se diagnostica y cuál es su tratamiento. * Artículo relacionado: "[Los 15 trastornos neurológicos más frecuentes](/clinica/trastornos-neurologicos-frecuentes)" ## Síndrome de Guillain-Barré: qué es y cómo se produce El síndrome de Guillain-Barré, o polirradiculoneuritis aguda, es una enfermedad neurológica rara, de origen autoinmune, que **se caracteriza por provocar un debilitamiento muscular rápido (de comienzo distal y avance proximal), acompañado de alteraciones en la sensibilidad**, como dolor o sensaciones de hormigueo y pérdida de reflejos osteotendinosos, pudiendo afectar también a la musculatura bulbar respiratoria. Este trastorno afecta principalmente al sistema nervioso periférico y **es la causa más frecuente de parálisis aguda generalizada**. El daño se produce en las [vainas de mielina](/neurociencias/mielina) de los nervios (que aumentan la velocidad de transmisión de los impulsos nerviosos), y es el propio sistema inmunológico del paciente el que lo provoca. El síndrome de Guillain-Barré afecta por igual a todas las razas, sexos y edades. Su incidencia es de 1 o 2 casos por cada 100.000 personas. El curso de la enfermedad puede ser fulminante, con una evolución rápida que suele requerir asistencia ventilatoria al cabo de pocos días. * Quizás te interese: "[Polineuropatías desmielinizantes: qué son, tipos, síntomas y tratamiento](/salud/polineuropatias-desmielinizantes)" ## Posibles causas Aunque aún se desconocen las causas, **las hipótesis más plausibles apuntan a un origen infeccioso de tipo vírico o bacteriano**, que podrían ser generadoras de una respuesta autoinmune que desencadena una reacción contra las proteínas básicas de los nervios, dando lugar al proceso de desmielinización. ## Diagnóstico **El síndrome de Guillain-Barré no puede diagnosticarse con la administración de una única prueba**. Se suele sospechar de su existencia cuando el paciente presenta los criterios diagnósticos de Asbury y Cornblath: una debilidad progresiva en más de un miembro y arreflexia osteotendinosa universal. Por otra parte, existen otra serie de rasgos clínicos que apoyan el diagnóstico; la progresión de la debilidad, que la afectación sea relativamente simétrica; que se presenten signos y síntomas sensitivos leves; que el paciente presente una disfunción autonómica (taquicardia, hipertensión arterial o signos vasomotores); que exista una afectación de los nervios craneales (con debilidad facial en la mitad de los casos); y la ausencia de fiebre. Aunque el cuadro clínico puede variar, el síndrome de Guillain-Barré **es la causa actual más común de debilidad simétrica que se desarrolla en apenas unas horas**. La parálisis progresiva, la insuficiencia respiratoria y las complicaciones cardiovasculares van a decantar el diagnóstico igualmente. Otras manifestaciones clínicas pueden variar de un paciente a otro, como por ejemplo: tener fiebre al comienzo; que se de una pérdida sensorial severa y con dolor; que cese la progresión de la enfermedad sin recuperación o con secuelas permanentes significativas; que los esfínteres se vean afectados; y **que haya lesiones en el sistema nervioso central**. El diagnóstico diferencial debe tener en cuenta los siguientes trastornos: enfermedades de las motoneuronas (como la poliomielitis viral aguda, la esclerosis lateral amiotrófica, etc.); polineuropatías (por ejemplo, la porfiria, otras formas del síndrome de Guillain-Barré, la enfermedad de Lyme, etc.); trastornos de la transmisión neuromuscular (como la miastenia gravis autoinmune o el botulismo); y otros trastornos musculares y metabólicos. ## Síntomas y signos clínicos Los síntomas iniciales en el síndrome de Guillain-Barré pueden implicar sensaciones anormales (parestesias) que se manifiestan de formas diversas, primero en una de las extremidades y más tarde en ambas, como por ejemplo: **hormigueos, entumecimientos, adormecimientos o sensación de tener algo que camina debajo de la piel (formicación)**. La debilidad muscular también está presente y suele iniciarse en los miembros inferiores, afectando después a otras zonas del cuerpo. Esta debilidad es, en ocasiones, progresiva y afecta a brazos, piernas, músculos respiratorios, etc., configurando el cuadro clínico típico del síndrome de Guillain-Barré. Los pares craneales también se ven afectados en un 25% de los pacientes, siendo la paresia facial bilateral el signo más característico. **La enfermedad sigue un curso que dura entre 3 y 6 meses, evolucionando en varias fases**: la fase de progresión, la estabilización y la recuperación o regresión. ### 1. Fase de progresión En la etapa de progresión, **la persona experimenta los primeros signos y síntomas como el hormigueo y la parestesia en pies y manos**, seguidas de la debilidad muscular que puede terminar en parálisis. Generalmente, suele comenzar en los pies o las piernas para después extenderse gradualmente al resto del cuerpo, provocando una parálisis facial o respiratoria. Esta primera fase puede durar desde unas horas hasta tres o cuatro semanas y, dependiendo de la gravedad de los síntomas, puede requerir intervención médica urgente, debido al posible bloqueo de las vías respiratorias. ### 2. Fase de estabilización Esta segunda etapa, conocida como fase de estabilización, **comprende el final de la progresión de la enfermedad y el inicio de la recuperación clínica**. En esta fase, los signos y síntomas del síndrome de Guillain-Barré suelen estabilizarse; no obstante, pueden aparecer problemas como la hipertensión o hipotensión, taquicardia y algunas complicaciones como las úlceras por presión, coágulos de sangre o infecciones de orina. La duración de la fase de estabilización es variable, pudiendo llegar a ser desde unos pocos días hasta varias semanas, o incluso meses. Con todo, cabe señalar que esta etapa puede estar ausente durante el curso de la enfermedad. ### 3. Fase de regresión o recuperación Esta última etapa está comprendida entre el inicio de la recuperación y el final de la enfermedad. Durante la misma, los síntomas van disminuyendo poco a poco. **A partir de esta última fase, si en el paciente persisten daños neurológicos ya pueden considerarse como secuelas permanentes**. Esta fase suele durar, aproximadamente, 4 semanas, aunque este tiempo varía de un sujeto a otro en función de la gravedad y extensión de las lesiones neurológicas, pudiendo alargarse durante meses. ## Tratamiento **Es bastante probable que el síndrome de Guillain-Barré curse con un rápido deterioro**, por lo que todos los pacientes de los que se sospeche la presencia de la enfermedad deben ser hospitalizados, y su función respiratoria ha de ser monitorizada. Del mismo modo, si el paciente presenta dificultades en la deglución, se le debe alimentar a través de una sonda estomacal. **En caso de que la persona presente parálisis respiratoria**, será necesaria la asistencia mediante aparatos de ventilación mecánica. El manejo de la función respiratoria incluye la permeabilidad de las vías aéreas, la capacidad de la persona para toser y expectorar, la habilidad para tragar y la aparición de síntomas de hipoxemia (disminución del oxígeno en sangre) o hipercapnia (aumento del dióxido de carbono en sangre). El tratamiento indicado para este trastorno incluye por un lado, la plasmaféresis, un procedimiento que consiste en depurar la sangre, esto es, extraer un volumen determinado de plasma sanguíneo para eliminar partículas y patógenos que intervienen en la respuesta inmune patológica; y por otro lado, la administración intravenosa de inmunoglobulinas, un tratamiento para sustituir las defensas de una persona cuando padece una enfermedad infecciosa o autoinmune. #### Referencias bibliográficas: * Hughes, R. A., & Cornblath, D. R. (2005). Guillain-barre syndrome. The Lancet, 366(9497), 1653 - 1666. * Tellería-Díaz, A., & Calzada-Sierra, D. J. (2002). Síndrome de Guillain-Barré. Rev Neurol, 34(10), 966 - 976.
Ver Fuente
Ver Fuente
0 notes
Text
ESCLEROSIS MÚLTIPLE
Epidemiología, etiología, patogenia
Definición.
Enfermedad inflamatoria crónica de etiología desconocida, caracterizada por la presencia de múltiples lesiones en la sustancia blanca del SNC, llamadas placas. Las placas son áreas de desmielinización formadas por un infiltrado de polimorfonucleares alrededor de las vénulas que, con el tiempo, da gliosis.
Es típica de adulto joven, lentamente progresiva, evoluciona en brotes con manifestaciones variables generalmente con remisiones y exacerbaciones y no tiene tratamiento.
Epidemiología.
· Es muy frecuente. La prevalencia en España es de 65 casos por cada 100.000 habitantes por año.
· Es la causa no traumática de invalidez más frecuente en el adulto joven (20-40 años).
· Es más frecuente:
n raza blanca
n occidente
n nivel socioeconómico alto
n clima templado
n área urbana
n mujeres
· Los países se dividen según la prevalencia: alta, media y baja.(hemisferio norte más prevalencia).
Etiología.
La causa es desconocida, pero se sabe que si una menor de 15 años de un país de baja prevalencia emigra a otro de alta, aumenta el riesgo de padecer la enfermedad y viceversa; pero si es mayor de 15 años conserva siempre el riesgo de su país de origen. Por tanto, parece que existen factores ambientales que determinan la susceptibilidad y que actúan antes de la pubertad. Se supone que son virus comunes extendidos ampliamente por las zonas de alta prevalencia aunque también se ha visto que puede existir relación con la dieta (se han encontrado títulos elevados frente a diversos virus como el sarampión).
Además existen razas con baja prevalencia (japoneses, gitanos, esquimales) por lo que deben existir factores genéticos.
Así que se combinan factores genéticos y ambientales que determinan la enfermedad ya que si un gemelo tiene esclerosis múltiple, el otro tiene un 30% de riesgo de tener la enfermedad y los padres del 1-2% de trasmitirlo a sus hijos. Estos porcentajes son mayores de los esperados si no existieran factores genéticos y menores si estuvieran determinados genéticamente.
La teoría que existe para explicarla es:
“Personas genéticamente predispuestas se infectan con un virus muy extendido en su país y esto causa la aparición de bien autoantígenos o bien antígenos virales en SNC que provocan una reacción inflamatoria. Las células sanguíneas van al SNC, reconocen el antígeno y liberan INF g, citoquinas y TNF que incrementan la permeabilidad de la BHE y permite que entren células inespecíficas (monocitos y macrófagos) que fagocitan la mielina. Con el tiempo la inflamación remite por citoquinas reguladoras (IL4, IL10, IL13) y células supresoras, pero en el SNC quedan células linfoides que aumentan las IgG del LCR y causan brotes de reactivación toda la vida.”
Esta teoría se basa en un modelo animal experimental, Encefalitis Anérgica Experimental que se induce al inyectar proteína derivada de la mielina a un animal. Aunque se han buscado antígenos específicos frente a la reactividad humoral y celular en el SNC, todavía no se han encontrado.
Anatomía patológica.
Macroscopía: Las lesiones patognomónicas son las PLACAS: son áreas de desmielinización grises o rosadas, nítidas, bien delimitadas, múltiples, asimétricas (en el tronco a veces son simétricas) de pequeño tamaño, y de localización aleatoria (con mayor afectación de la región periventricular, nervio óptico, tronco del encéfalo, cerebelo y médula espinal). Siempre en SNC y nunca en SNP. Se encuentran en número mayor de lo esperado por la clínica porque existen áreas silentes.
Microscopía: La placa reciente tiene un centro con infiltrado perivenular de CD4, CD8, macrófago, microglía y astrocitos y una zona con desmielinización y axones conservados. La placa vieja tiene reacción glial con astrocitos y no tiene células inflamatorias.
El número de oligodentrocitos, productores de la mielina en el SNC, es normal o alto en el borde y bajo en el interior por lo que tiene durante cierto tiempo una regeneración limitada (placas en sombra) antes de morir (puede explicar las remisiones y exacerbaciones de la enfermedad). Los axones al principio están bien pero con el tiempo se lesionan y el defecto se vuelve irreversible; se detecta por TAC y SPECT.
Existen unas variantes:
- Variante hiperaguda de Marburg que es muy grave, puede ser mortal y tiene muchos macrófagos y reacción periaxonal con edema.
- Variante de Baló desmielinización en capas concéntricas
- Esclerosis difusa de Schilder.
- Neuromielitis óptica de Devic: afectación del nervio óptico que da muchas secuelas, a veces necrosis y cavitación y es secundaria a esclerosis múltiple y también a mielitis.
Fisiopatología
La sustancia blanca es un aislante que preserva la onda de despolarización de un nodo de Ranvier a otro (conducción saltatoria), y aumenta la velocidad de conducción. La desmielinización que asienta en áreas silentes del encéfalo no da clínica, pero si asienta en áreas activas da sintomatología por disminución de la velocidad o bloqueo de la conducción, y al evolucionar, puede ser reversible, pero si daña el axón es irreversible.
Los enfermos son muy sensibles a:
· Variación de la composición iónica (existen fármacos que interfieren con canales de potasio para mejorar la conducción)
· Dolor
· Incremento de la temperatura
Fenómeno de Uhthoff: Tras una mielitis óptica, al hacer ejercicio, aparece una amaurosis unilateral transitoria que desaparece con el reposo y lleva a una claudicación del nervio óptico.
Clínica
La tercera década de la vida es la de mayor incidencia; Luego aparece un segundo pico 40-35 años.
Dos mujeres por cada hombre.
Manifestaciones clínicas variables: es una enfermedad crónica con curso creciente y progresivo. El inicio suele ser insidioso.
Manifestaciones motoras:
Determinan el pronóstico.
Puede ser:
n Remitente: tiene brotes que dan déficit motor de la vía piramidal y debilidad principalmente en miembros inferiores (la forma más frecuente)
n Crónica progresiva
Sufre disminución de fuerza, aumento de reflejos, Babinski positivo.
Remite cada brote pero va dejando secuelas que se van sumando y con el tiempo disminuye la fuerza de los miembros inferiores y al final parálisis (silla de ruedas).
Fatiga: sensación de cansancio desproporcionada por aumento de la conducción nerviosa (mejora con amantadina)
Manifestaciones sensitivas:
Son muy frecuentes.
- Positivas: parestesias, disestesias, dolor (principalmente neuralgia del trigémino)
- Negativas: la disminución de la sensibilidad es principalmente el cordón posterior (vibratoria y posicional)
- Signo de Lhermitte: sensación de descarga eléctrica que baja por la zona central de la espalda a los miembros inferiores al flexionar el cuello.
Manifestaciones visuales:
- Neuritis óptica (retrobulbar o de la papila): dolor retrocular que progresa a una disminución de la visión de borroso a ceguera. Evoluciona favorablemente aunque a veces queda ceguera parcial.
- Signo de Marcus Gunn: neuritis óptica de un lado que al estimular con la luz el ojo sano el enfermo se contrae y si pasas la luz al enfermo también se contrae.
- Oftalmoplejia transitoria con diplopía: lesión del fascículo longitudinal medial del tronco, que hace que un ojo no pase la línea media y el sano sí pero presenta nistagmus.
Manifestaciones cerebelosas:
Éstos y los piramidales tienen mal pronóstico.
Presenta ataxia de la marcha (como borracho), dismetría, temblor de intención, disartria (lenguaje escandido; es decir, enunciación lenta con tendencia a dudar al inicio de una palabra o sílaba. Frecuente en edad avanzada). Generalmente se manifiesta la triada de Charcot (nistagmo, temblor de intención y lenguaje escandido).
Normalmente no es clínica cerebelosa pura.
Manifestaciones por alteración del tronco:
Presenta oftalmoplejia interneural, parálisis facial, vértigo (frecuente), sordera (rara).
Manifestaciones de los esfínteres:
Urgencia urinaria, pérdida del control esfinteriano, impotencia, dificultad coital por disminución de la lubricación y sensibilidad perineal.
Normalmente afecta a esfínter vesical y raramente al rectal.
Manifestaciones por alteraciones de las funciones superiores:
Alteración de la concentración y del rendimiento mental, frecuente la habilidad emocional, y a veces demencia franca con rasgos subcorticales. Puede haber crisis convulsivas.
Fenómenos paroxísticos: aparición de ataxia, disartria, crisis tónicas, disminución de la fuerzas, acinesia… normalmente doloroso que dura segundos (tratamiento: carbamacepina).
Evolución.
Impredecible. Varía dependiendo dela evolución del enfermo a los 5-10 años. Existen varias formas:
n Remitente recidivante: los brotes remiten y tras un período variado reaparecen igual o distinto. El periodo interbrotes provoca secuelas que van sumándose. Suele durar unos 25 años.
n Crónica progresiva: deterioro progresivo. A veces es mortal en el plazo de 1 año.
n Formas intermedias: con brotes y progresión en las intercrisis.
n Formas benignas: con brotes que para a los 10-20 años. Tras 10 años de brotes tienen más secuelas.
A veces siguiendo a los pacientes con RMN, se ve que aparecen y desaparecen placas sin dar clínica (zonas silentes).
Es más frecuente la evolución benigna.
Son signos de mal pronóstico la progresión desde el principio, edad mayor de 40 años y síntomas piramidales aunque no se puede dar un pronóstico hasta los 5-10 años de evolución.
Diagnóstico.
Es clínico por los criterios de Poser. Sin embargo, generalmente no puede establecerse el diagnóstico de certeza durante el primer episodio, aun cuando se sospeche. Posteriormente una historia de remisiones con signos clínicos de lesiones del SNC diseminadas es altamente sugestiva.
Pruebas accesorias:
· LCR (anormal en más del 55% de los casos):
- presencia intratecal de IgG:
índice IgG = IgGLCR x AlbSUERO / AlbLCR x IgGSUERO ³ 0.70
- con electroforesis o isoenfoque se ven bandas oligoclonales IgG (indican una síntesis de IgG en el interior de la BHE) (90% de los casos).
- pleocitosis menor de 50-100 células.
- glucosa normal.
- proteínas altas por aumento de la permeabilidad de BHE.
· Potenciales evocados somatosensoriales, auditivos y visuales; retraso en la conducción. Lo más importante es si un paciente asintomático tiene potencial visual asimétrico.
· RMN: lesiones múltiples en T2 en sustancia blanca periventricular, cuerpo calloso, tronco, médula, fosa posterior y nervio óptico. El borde de la lesión capta gadolinio. Especial atención al área del agujero occipital.
Diagnóstico diferencial:
Infartos cerebrales pequeños, siringomielia, ELA, sífilis, enfermedad de Lyme, HTLV1 (paraparesia espástica tropical), SIDA, LES, sarcoidosis, síndrome de Sjögren, enfermedad de la mielina, déficit de B12, a veces tumores, ataxias hereditarias …
Tratamiento: NO EXISTE TRATAMIENTO ESPECÍFICO.
El paciente debe mantener una vida lo más normal y activa posible, pero evitando el exceso de trabajo y la fatiga. Debe evitarse la falta de esperanza.
- Tratamiento sintomático para:
* alteración cerebelosa.
* espasticidad piramidal: masaje y movimientos pasivos (entrenamiento muscular); Baclofeno 100mg/d en bomba de infusión en espacio subaracnoideo reduce la espasticidad inhibiendo los reflejos de la médula espinal (ojo, porque exacerba a menudo la debilidad, incapacitando con ello aun más al paciente).
* alteración esfinterial: tratamiento inmediato de las infecciones y dificultades urinarias.
- Tratamiento de los brotes: esteroides artificiales iv 1g/d hasta que las manifestaciones remitan (durante unos 4d), seguida de una disminución de las dosis hasta la supresión en 5-7 días; efectos evidentes especialmente si los fármacos se administran al principio del episodio. Antes se usaba ACTH.
- Tratamiento para limitar la progresión de la enfermedad:
· Inmunodepresores:
a) Azatioprina 150 mg/d
b) Ciclofosfamida
· Inmunomoduladores:
INFb 8 x 106 U subcutánea a días alternos (para pacientes con dos brotes en los dos últimos años sin alteración neurológica en periodos interbrote). No debe considerarse como tratamiento curativo pero puede alterar la historia natural de la enfermedad (IMPORTANTE).
0 notes
Text
Investigadores descubren un nuevo subtipo de esclerosis múltiple
Investigadores descubren un nuevo subtipo de esclerosis múltiple
EUROPA PRESS
Presenta pérdida neuronal pero no desmielinización de la sustancia blanca del cerebro.
Investigadores de la Cleveland Clinic (Estados Unidos) han descubierto un nuevo subtipo de esclerosis múltiple (EM) que presenta pérdida neuronal pero no desmielinización de la sustancia blanca del cerebro.
La esclerosis múltipleha caracterizado durante mucho tiempo como una enfermedad de la…
View On WordPress
0 notes
Text
¿Qué tengo?
Inició en mayo del año pasado cuando me percaté de una molestia en las articulaciones de los dedos de manos y pies aunado a una sensación de ardor en las yemas que se acrecentaba al contacto.
El primer diagnóstico fué Rye, debido a mi consumo de aspirinas pero al suprimir su uso, las molestias continuaron incrementándose lo que nos llevó a una posible artrosis ocupacional. Luego de análisis clínicos surgió el posible diagnóstico de neuropatía periférica con complicaciones ocasionada por un factor aún no identificado
Luego de una crisis nerviosa aguda resulté con un debilitamiento general, manos frías y las extremidades casi inútiles; me fue casi imposible incorporar y mucho menos pasear a Frida. Luego recuperé las fuerzas lenta y lastimosamente (huelga decir que perdí mi trabajo).
El ardor en las yemas se incrementó junto a un dolor casi insoportable en las palmas y tobillos de los pies y muñecas que no pocas veces me arrancó angustiantes lágrimas de desesperación e insomnio.
Tramadol fue una parcial victoria pues si bien ahora puedo dormir, los dolores articulares continúan y tengo que administrarlo tan seguido que, sumado al consumo masivo de aspirinas y otros analgésicos, me orillan a temer por la integridad de hígado y riñones.
La frustración continúa al escuchar de mi doctor que mi padecimiento es autoinmune y crónico; una posible fibromialgia para la que no hay tratamientos y que deberé aceptar ese dolor incapacitante por el resto de mi vida, lo cual me niego a aceptar.
La situación se complica enormemente al carecer de trabajo y de seguridad social amén que en mi familia sospechan que es psicosomático (¡seguramente finjo para no sé qué chingaos!). No tengo de momento perspectivas de trabajo, amor o incluso fraternidad ni mucho menos independencia económica.
Llevo más de un año de casi completo aislamiento social (ni mis mejores amigos están al tanto). Estos dolores tan intensos que van y vienen me dificultan enormemente escribir en la PC o incluso en mi celular (donde escribo de a dedito). Casi no puedo manipular los controles de mis cámaras y mucho menos tocar la guitarra. Así las cosas; trato de no agüitarme y ver luminoso el mañana. Continuaré buscando respuestas en otras opiniones y otros diagnósticos.
Actualización Jul 2018
Luego de un rápido examen mi Doctora mencionó la palabra: "desmielinización", restando saber qué la causa y un posible tratamiento.
0 notes
Text
Cuando se debe iniciar, cambiar o suspender el tratamiento en la esclerosis múltiple.Revisión 2018.
La Academia Americana de Neurología (AAN) publicó en la revista Neurology el 23 abril 2018 las recomendaciones para iniciar, cambiar y suspender las terapias modificadoras de la enfermedad (DMT) relevantes para los pacientes con:
EM remitente-recurrente (EMRR),
EM secundaria progresiva,
EM progresiva primaria (EMPP)
y síndromes clínicamente aislados de desmielinización.
Tratamientos modificadores…
View On WordPress
0 notes
Video
youtube
NeoNervial Soluciones al dolor por Io3M Instituto de Ozonoterapia Médica
¿Qué es NEONERVIAL® ?
Complemento alimenticio que aporta los nutrientes esenciales para la formación de la mielina (envoltura o vaina que recubre los nervios). La vaina de mielina permite que los impulsos eléctricos se transmitan de manera rápida y eficiente a lo largo de las neuronas.
NUEVA FORMULA CON FUNCIONES NEUROPROTECTORAS
¿COMO ACTUA?
NEONERVIAL® actúa de forma rápida y eficaz favoreciendo la síntesis de los fosfolípidos y glucolípidos que integran la vaina de mielina y otras estructuras nerviosas. NEONERVIAL® desencadena una intensa actividad metabólica que facilita, a su vez, el proceso de regeneración de la vaina de mielina (envoltura de los nervios), corrigiéndose así la desmielinización que se produce en las lesiones nerviosas.
0 notes
Photo



LA RATA TAIEP: ESTRELLA DE LAS PASARELAS PARA EL ESTUDIO DE TRATAMIENTOS CONTRA LA ESCLEROSIS MÚLTIPLE · Evaluarán los efectos de tratamientos farmacológicos y fisioterapéuticos en el andar de roedores con este mal neuronal BUAP. 16 de mayo de 2017.- La rata TAIEP es una estrella de la comunidad científica y las pasarelas. Este modelo animal, desarrollado en el Instituto de Fisiología (IF) de la BUAP, ha mostrado sus bondades en el estudio de padecimientos asociados al cerebro, ya que presenta temblor, ataxia, inmovilidad, epilepsia y parálisis, síntomas que le dan su nombre. En una reciente investigación, este roedor se luce por una pasarela para que los especialistas valoren el efecto de fármacos y la fisioterapia en el tratamiento de la esclerosis múltiple. Pero la rata TAIEP no camina por una pasarela tradicional, se trata del CatWalk de la compañía Noldus: una estructura con forma de pasillo flanqueada con barreras que permite observar a detalle la locomoción del roedor, la velocidad al andar, la coordinación entre las extremidades y su equilibrio, entre otros aspectos. Estas variables permiten hacer una evaluación integral de la marcha e identificar los progresos de distintos tratamientos para la esclerosis múltiple, que según la prensa nacional, afecta a casi 20 mil mexicanos, provocándoles disfunciones en el cerebro, nervios ópticos y en la médula espinal. María del Carmen Cortés Sánchez y José Ramón Eguibar Cuenca, investigadores adscritos al Laboratorio de Neurofisiología de la Conducta y Control Motor del IF-BUAP, son los responsables de este proyecto de investigación. Este último destacó que la rata TAIEP es un modelo animal único e idóneo para el estudio de enfermedades neuronales, porque es un modelo genético que desarrolla la enfermedad tal y como un paciente humano lo hace durante su vida. “Los ejemplares caminan con ataxia –dificultad para coordinar movimientos-al desplazarse de un punto a otro. Este aparato –el CatWalk- nos permite ver las huellas de las patas de la ratas, tomar la velocidad con la que avanzan, conocer cómo abren el ángulo de sostén de las extremidades, detectar cuál de estas presionan más o cómo mantienen el balance a pesar de sus problemas motores”, señaló el especialista. A diferencia de las fotografías que capturan los mejores fotógrafos de moda, que buscan reflejar lo mejor de las marchas en las pasarelas, en este proyecto, con la información obtenida del CatWalk, se busca evidenciar con claridad y detalle las dificultades que al andar padecen los roedores. Una vez descrita la marcha, los científicos de la BUAP, en colaboración con el doctor Hugo Hernández y la alumna de Ingeniería Biomédica Saraí Domínguez, investigadores de la Universidad de Guanajuato, administrarán fármacos a nivel espinal –vía la cisterna magna- para observar las modificaciones en su andar y, de esta forma, evaluar si existen mejoras en la locomoción de la rata TAIEP. Los fármacos suministrados son factores de crecimiento neuronal de efectos conocidos en otros trastornos cerebrales. “Lo que queremos es mejorar la comunicación de las neuronas, es decir, su sinapsis. La rata TAIEP presenta estos problemas debido a deficiencias con la mielina, sustancia que recubre las vías de comunicación entre las células del cerebro (neuronas), tal y como el plástico aísla los cables de cobre en las conexiones de nuestra casa”, comentó Cortés Sánchez. Si las instalaciones eléctricas no cuentan con esta protección se ocasionan cortos circuitos; algo similar sucede en las comunicaciones entre distintas estructuras cerebrales y en particular en el circuito de la marcha. La rata TAIEP pierde la mielina con la edad (desmielinización), lo que altera su marcha de manera progresiva y la convierte en un modelo ideal para estudiar enfermedades asociadas con ésta, como lo es la esclerosis múltiple o la esclerosis lateral amiotrófica, conocida también como enfermedad de Lou Gehring. Un brazo robótico para la rehabilitación El progreso de pacientes con esclerosis múltiple se debe en parte a los tratamientos fisioterapéuticos, ya que reactivan partes del cerebro asociadas al control motor que están dañadas por el mal congénito. A la fecha, éstos son realizados de forma manual por especialistas médicos. Científicos del Instituto de Fisiología y la Facultad de Ciencias de la Electrónica (FCE) proponen su automatización, es decir, trabajan en el diseño y fabricación de un brazo robótico que realice las terapias que los fisioterapeutas ejecutan de forma manual. El prototipo será sometido a prueba en el tratamiento de las ratas TAIEP. Sus resultados serán evaluados de la misma manera con la que se mide el impacto de los factores de crecimiento neuronal. Lo anterior permitirá conocer los efectos benéficos de la terapia automatizada y combinarla con otras opciones terapéuticas, como los tratamientos farmacológicos o el factor de crecimiento neuronal. En la opinión de Fernando Reyes Cortés, especialista en automatización y robótica de la FCE y asesor del área del diseño del robot en cuestión, las ventajas de la automatización se deben a que se reduce el margen de error durante las terapias: “los movimientos que deben ser repetidos serán idénticos tanto en intensidad, velocidad, dirección y trayectoria”, y a la implementación a larga distancia de dichos tratamientos, con la ayuda de dispositivos móviles. El investigador describió al prototipo como un robot con peso de 10 a 8 veces superior al de los roedores –para evitar que estos lo muevan al interactuar-, el cual está integrado por una base móvil –una suerte de carrito- que soporta un brazo robótico que se encargará de hacer los movimientos a las extremidades afectadas. Este último componente es una escala del 30 por ciento del brazo robótico diseñado por el mismo investigador y su equipo de trabajo, para atender problemas de movimiento en humanos. El diseño, fabricación y calibración del robot se realiza a partir de los parámetros que se obtengan al analizar la marcha del roedor en el CatWalk. Los roedores al caminar dejarán sus huellas en este dispositivo especializado, para analizar con detalle la disposición de las extremidades, la relación que guardan entre ellas y la posición de los dedos.
0 notes
Text
Polineuropatías desmielinizantes: qué son, tipos, síntomas y tratamiento
**Las polineuropatías desmielinizantes son un conjunto de trastornos que afectan al sistema nervioso** y producen alteraciones en las funciones motoras y sensitivas. Su principal característica es la pérdida de mielina que se produce en las células nerviosas y que es responsable de los problemas que presentan estos pacientes. A continuación, te explicamos en qué consisten y cuáles son las características de este tipo de trastornos, cómo se diagnostican, cuáles son los principales tipos que existen y el tratamiento actual disponible. * Artículo relacionado: "[Mielina: definición, funciones y características](/neurociencias/mielina)" ## Polineuropatía desmielinizante: definición y características Las polineuropatías desmielinizantes son un grupo de enfermedades neurológicas, que pueden ser hereditarias y adquiridas, **caracterizadas por provocar daños en la mielina de las fibras nerviosas del Sistema Nervioso Periférico**. Generalmente, este tipo de trastornos cursan con disminución o pérdida de la fuerza muscular y/o pérdida sensitiva. La desmielinización es un proceso que implica una pérdida o un daño en la vaina de mielina que recubre los axones de las células nerviosas. La principal función de la mielina es aumentar la velocidad de transmisión de los impulsos nerviosos, por lo que es fundamental para que funcione correctamente la actividad del sistema nervioso. **Las patologías que cursan con desmielinización suelen afectar a funciones básicas** y repercuten de forma significativa en la vida de los pacientes. Las alteraciones pueden ser desde problemas musculares o sensitivos, hasta el deterioro cognitivo y funcional que pueda incapacitar a la persona de forma permanente y absoluta. ## Diagnóstico Los trastornos desmielinizantes que afectan a los nervios periféricos se suelen diagnosticar basándose en la observación de los síntomas y los signos, tras realizar pruebas electromiográficas (que evalúan el estado de los músculos y los nervios), estudios genéticos y, en ocasiones, datos recogidos de la biopsia del nervio. Para poder diagnosticar correctamente una polineuropatía desmielinizante, **se debe diferenciar esta enfermedad de otro tipo de polineuropatías y trastornos que también afectan al sistema nervioso periférico** (como las mononeuropatías, radiculopatías, etc.), y se ha de establecer el mecanismo que ha provocado el daño (desmielinizante o axonal), así como la causa de la enfermedad. Durante la recogida de datos y la realización del diagnóstico, se deben considerar otros aspectos relevantes como: la modalidad de afectación (predominio sensitivo, motor, etc.), los tipos de fibras afectadas (gruesas o finas), el perfil temporal (agudo, subagudo o crónico), el perfil evolutivo (monofásico, progresivo o recidivante), la edad de inicio, presencia o ausencia de tóxicos, antecedentes familiares y la existencia de otros trastornos concurrentes. ## Tipos Existen múltiples variantes de polineuropatías desmielinizantes y su clasificación más habitual se realiza en base a un criterio de origen; es decir, en si son de tipo hereditario o adquirido. Veamos cuáles son: ### 1. Hereditarias Las polineuropatías desmielinizantes hereditarias **están asociadas a defectos genéticos específicos**, a pesar de que aún se desconocen cuáles son los mecanismos a través de los cuales dichas mutaciones causan las manifestaciones patológicas de desmielinización. Existen multitud de variantes hereditarias de este trastorno. Aquí haremos un repaso a tres de ellas: la enfermedad de Charcot-Marie-Tooth, la enfermedad de Refsum y la leucodistrofia metacromática. Veamos cuáles son sus principales características y manifestaciones clínicas. #### 1.1. La enfermedad de Charcot-Marie-Tooth Existen más de 90 variantes de esta polineuropatía hereditaria y cada tipo es causado por distintas mutaciones genéticas. La enfermedad de Charcot-Marie-Tooth afecta por igual a todas las personas, razas y grupos étnicos, y en el mundo la sufren cerca de 2,8 millones de personas. En los tipos más comunes, los síntomas suelen comenzar a los 20 años de edad y pueden incluir: deformidad del pie, incapacidad para mantener el pie horizontalmente, los pies suelen golpear el suelo al caminar, pérdida muscular entre las piernas, entumecimiento de los pies y problemas de equilibrio. También pueden aparecer síntomas similares en brazos y manos, y **la enfermedad raramente afecta a la función cerebral**. #### 1.2. Enfermedad de Refsum La enfermedad de Refsum **es una neuropatía hereditaria sensitivo-motora que se caracteriza por la acumulación de ácido fitánico**. Su prevalencia es de 1 persona por cada millón, y afecta por igual a hombres que a mujeres. Los síntomas iniciales suelen originarse alrededor de los 15 años de edad, aunque pueden aparecer también durante la infancia o en la edad adulta (entre los 30 y 40 años). La acumulación de ácido fitánico provoca en los pacientes lesiones en la retina, el cerebro y el sistema nervioso periférico. En la mayoría de los casos, la causa de este trastorno es una mutación en el gen PHYN, aunque estudios recientes han descubierto que otra posible mutación, en el gen PEX7, podría ser también un factor causal. #### 1.3. Leucodistrofia metacromática La leucodistrofia metacromática es una enfermedad neurodegenerativa caracterizada por **la acumulación de sulfátidos en el sistema nervioso central y los riñones**. Existen tres tipos: la infantil tardía, juvenil y adulta. La prevalencia de este trastorno se estima en torno a 1 caso de cada 625.000 personas. La forma infantil tardía es la más habitual y suele comenzar a edades en las que los niños aprenden a caminar, con síntomas como hipotonía, dificultades para andar, atrofia óptica y regresión motora que precede al deterioro cognitivo. El sistema nervioso periférico de estos pacientes resulta sistemáticamente dañado (disminuye drásticamente la velocidad de conducción nerviosa). * Quizás te interese: "[Los 15 trastornos neurológicos más frecuentes](/clinica/trastornos-neurologicos-frecuentes)" ### 2. Adquiridas Las polineuropatías desmielinizantes de tipo adquirido **representan un grupo heterogéneo, con multitud de tipos y variantes**. Estas enfermedades pueden tener distintas causas: tóxicas (como metales pesados), por carencias (de vitamina b12, por ejemplo), metabólicas, inflamatorias o infecciosas, inmunitarias, entre otras. La polineuropatía inflamatoria desmielinizante crónica (CIDP) constituye una de las formas más comunes de este tipo de polineuropatías, y una de sus variantes más conocidas es la enfermedad o síndrome de Guillain-Barré. A continuación, veremos cuáles son sus principales características y manifestaciones clínicas. #### 2.1. Polineuropatía inflamatoria desmielinizante crónica (CIDP) La CIDP es, como decíamos, una de las formas más comunes de polineuropatías adquiridas. **Se inicia de forma insidiosa y suele progresar durante, al menos, 2 meses**. Su curso puede ser recurrente o crónicamente progresivo, y generalmente es de predominio motor, afectando a grupos de músculos proximales y distales. Esta enfermedad tiene una incidencia de 0,56 casos por cada 100.000 personas. La edad media de aparición del trastorno está en torno a 47 años, aunque incide en todos los grupos etarios. Las manifestaciones clínicas de esta polineuropatía incluyen debilidad muscular proximal y pérdida de la sensibilidad distal en las extremidades que son progresivas y simétricas. Además, esta enfermedad **suele cursar con una disminución o, en ocasiones, la pérdida total de los reflejos osteotendinosos**. Aunque existen variantes con afectación puramente motora, son las menos frecuentes (en torno al 10% de los casos). Los nervios craneales no suelen verse afectados y un síntoma habitual suele ser la paresia del nervio facial bilateral. De forma infrecuente, también se ve afectada la capacidad respiratoria y la micción. #### 2.2. El síndrome de Guillain-Barré El síndrome de Guillain-Barré, también conocido como polineuropatía idiopática aguda, es un trastorno que provoca la inflamación de los nervios periféricos. Se caracteriza por una **aparición brusca de debilidad muscular y, a menudo, parálisis en las piernas, brazos, músculos respiratorios y la cara**. Esta debilidad se acompaña, frecuentemente, con sensaciones anómalas y la pérdida del reflejo rotuliano. La enfermedad puede manifestarse a cualquier edad y en personas de todas las etnias y lugares. Aunque se desconocen las causas de esta enfermedad, en la mitad de los casos se presenta después de una infección viral o bacteriana. Las investigaciones actuales sugieren que podría haber un mecanismo autoinmune responsable del proceso de desmielinización que caracteriza a este trastorno. ## Tratamiento El tratamiento indicado **varía en función del tipo de polineuropatía desmielinizante y de sus síntomas y manifestaciones clínicas**. En el caso de la CIDP, el tratamiento suele incluir corticosteroides como la prednisona, que puede prescribirse sola o en combinación con medicamentos inmunodepresores. También existen otros métodos terapéuticos eficaces, como por ejemplo: la plasmaféresis o intercambio plasmático, un método mediante el cual se extrae la sangre del cuerpo del paciente y se procesan los glóbulos blancos, los glóbulos rojos y las plaquetas, separándolas del resto del plasma, para luego reintroducirlos en la sangre; y la terapia con inmunoglobulina intravenosa, que suele utilizarse para tratar enfermedades que provocan inmunodeficiencia, y también en terapias inmunomoduladoras. Por otra parte, **la fisioterapia también puede resultar útil** en pacientes que sufren neuropatías desmielinizantes, ya que puede mejorar la fuerza, la función y la movilidad muscular, así como minimizar los problemas en músculos, tendones y articulaciones que suelen padecer este tipo de pacientes. #### Referencias bibliográficas: * Ardila, A., & Rosselli, M. (2007). Neuropsicología clínica. Editorial El Manual Moderno. * González-Quevedo, A. (1999). Polineuropatía crónica desmielinizante inflamatoria: una contribución a la caracterización de la enfermedad. Rev Neurol, 28(8), 772 - 778.
Ver Fuente
Ver Fuente
0 notes
Last Seen Blogs

hotanddistraught
game quinning goal!

a-duck-of-wellington
A Duck

ilovemesomevincentprice
Ramblings of a Vincent Price fangirl...

kirika-namikaze
kirika_namikaze

zombie-katsumi
tangöze