#Holoprosencephaly
Text
Happy disability pride month to people with brain malformations btw.
#I have a chairi malformation#no one ever talks abt us#disability pride month#ACC#septo-optic dysplasia#Holoprosencephaly#Lissencephaly#chiari malformation#schizencephaly#hemimegalencephaly#Rhomboencephalosynapsis#cripple punk
133 notes
·
View notes
Text
What do Sonic the Hedgehog and cyclopia have to do with each other? A lot, actually.
Alobar holoprosencephaly (aka cyclopia) is a condition in which the prosencephalon does not properly split during development. Let me explain what that means. So, one of the most important structures in the early embryo is the neural tube. This tube comes from the ectoderm and eventually becomes the central nervous system. The top of it has three little bulbs and the bottom is the spinal cord.
The top bulb is the prosencephalon, which will later differentiate into the telencephalon and diencephalon. These will become the cerebral hemispheres, basal ganglia, thalamus, hypothalamus, and amygdala. The middle bulb is the mesencephalon, which will become the midbrain. The bottom bulb is the rhombencephalon, which will differentiate into the metencephalon and myelencephalon. These will become the pons, cerebellum, and medulla.
So, this entire thing has to split in half anteriorly. The anterior portion of this structure deals with motor function, and the posterior half deals with sensory function. This holds true as an adult. The front half of your spinal cord carries motor nerves, and the back half carries sensory info. If you want to move your eyes, you use the front part of your brain. If you want to know what you are seeing, you use the back half.
If you look at your spinal cord, the front half has this fissure down the middle (anterior median fissure is such a fitting name, then right??). Your brain is also split down the middle. Since the eyes are part of your brain, you get one for each half of the brain (two). Now, let's say you never have this anterior fissure form... you won't get two eyes, either. The condition where the fissure doesn't form is called holoprosencephaly (holo means whole).
There are five types of this (in order of decreasing severity): alobar, semilobar, lobar, syntelencephaly, and microform.
*note that the more severe the condition is, the more likely the fetus will die and not form all structures mentioned*
Alobar: there will be one eye, a proboscis (fleshy tube on the face), and severe mental impairment.
Semilobar: eyes will be close together, the nasal bridge will be flat, the palate and lip will be cleft, and there will be mental and ocular impairment.
Lobar: the eyes will be close together, the nasal bridge will be flat, the nostrils will be close together, and there will be some mental impairments.
Syntelencephaly: flat nasal bridge, shallow philtrum, and possible mental delays.
Microform: eyes may be close, nasal bridge will be sharp, and there will be one central upper incisor.
Okay, so what does this have to do with Sonic the Hedgehog? Sonic hedgehog (SHH) genes, of course! This gene codes for the sonic hedgehog protein, which controls what happens during organogenesis (how everything differentiates and forms). A mutation in this gene can cause issues with how embryos form, and is indicated specifically in holoprosencephaly. SHH genes are still important in adults, and mutations can be linked to certain cancers.
Fun fact: an inhibitor of SHH genes is called Robotnikinin. I personally love this. And, here's a citation in case you don't believe me: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2770933/
#med student#medical school#medicine#biology#med school#med studyblr#anatomy#mcat#usmle#embryology#holoprosencephaly#cyclopia#cyclops#sonic hedgehog#sonic hedgehog genes#robotnikinin#brain stuff#neurology
3 notes
·
View notes
Text
Holoprosencephaly_Sound therapy session_Sounds of nature

Holoprosencephaly is a rare and complex neurological disorder characterized by abnormal brain development. Traditional medical treatments for Holoprosencephaly focus on managing symptoms and supporting overall development. However, recent research suggests that incorporating resonant frequency sound therapy as an adjunctive treatment can provide additional benefits. Resonant frequency sound therapy is a non-invasive therapeutic approach that utilizes specific low-frequency vibrations to stimulate the body's natural healing processes. It is based on the principle that every organ and tissue in our body emits its own unique resonant frequency. By introducing carefully calibrated sound waves, resonant frequency sound therapy aims to restore balance and promote optimal functioning. Resonant frequency sound therapy has the potential to stimulate neurological pathways and enhance brain function. By introducing therapeutic vibrations, this therapy may help activate dormant areas of the brain affected by Holoprosencephaly. Neurological stimulation can facilitate neural connections, potentially improving cognitive function, motor skills, and overall neurological development. Holoprosencephaly can be emotionally and physically challenging for both individuals and their families. Resonant frequency sound therapy has shown remarkable potential in inducing deep relaxation, reducing stress, and promoting a sense of calmness. By alleviating emotional and physical stress, this therapy may support overall well-being and improve the quality of life for individuals with Holoprosencephaly. Many individuals with Holoprosencephaly experience sensory processing difficulties. Resonant frequency sound therapy can aid in sensory integration by providing gentle and consistent sensory input. These therapeutic vibrations can help individuals regulate their sensory experiences, potentially improving their ability to process and respond to sensory stimuli. Resonant frequency sound therapy offers a holistic approach to the treatment of Holoprosencephaly. By addressing not only the physical symptoms but also the emotional and energetic aspects of the individual, this therapy complements traditional medicine and supports a comprehensive treatment plan. The combined approach can provide a more well-rounded and multidimensional approach to managing Holoprosencephaly. Resonant frequency sound therapy has the potential to improve overall well-being for individuals with Holoprosencephaly. The therapeutic vibrations can promote relaxation, reduce pain and discomfort, and enhance emotional and mental well-being. By fostering a state of balance and harmony within the body, this therapy may contribute to a higher quality of life for both individuals and their families. Resonant frequency sound therapy shows promise as an adjunctive treatment for individuals with Holoprosencephaly. By providing neurological stimulation, promoting relaxation, supporting sensory integration, and offering a holistic approach, this therapy can complement traditional medical treatments and contribute to a more comprehensive treatment plan. TO ACHIEVE A POSITIVE RESULT, DAILY LISTENING TO VIDEOS IS REQUIRED. I wish you health and prosperity!
#resonantfrequencytherapy#Holoprosencephaly#adjunctivetreatment#traditionalmedicine#benefits#braindevelopment#neurologicaldisorder#holisticapproach#therapeuticvibrations
0 notes
Text

My 25 years of palaeoart chronology...
A 2010 sketch of a baby therizinosaur with cyclopia (alobar holoprosencephaly), a congenital disorder characterised by one eye.
#Art#Painting#PaleoArt#PalaeoArt#SciArt#SciComm#DigitalArt#Illustration#Dinosaurs#Birds#Reptiles#Palaeontology#Paleontology#JurassicPark#JurassicWorld
75 notes
·
View notes
Note
Hi! First off I'd like to say I've learned a lot from this blog and I appreciate what you all do.
I was wondering what your opinion/opinions are on non-human characters who are intellectual disabled. I've been reading through your intellectual disability posts that you already have as well as doing research on some specific causes. Since they often seem to have a genetic component I imagine it would work differently for a species that doesn't reproduce in the same way humans do? What I'm specifically wondering about is if you would prefer a parallel to actual real life causes of intellectual disability or name a condition that already exists? Or something else?
Hi, I'm glad you enjoy the blog !
In my opinion, both would be fine. If you have (example) an anthro cat who's intellectually disabled, I don't think it there would be much difference between “strongly implying that they have feline Down syndrome via them being ID and having some DS-coded facial features” and “openly stating they have Down syndrome”, even though cats can't have it by definition.
You can also make the cause up if you're talking about fantasy/alien/non-existent species - maybe your character's disability could be caused by partial monosomy 98 because they happen to have way more chromosomes than humans do. In this case, you could pull some inspiration from the human counterparts - Angelman syndrome, Distal 18q-, etc.
Other genetic conditions like Rett syndrome, or Tuberous sclerosis, or all the X-linked causes could also probably be either named or just strongly alluded to. Unless the species has some very different anatomy, they would probably present the same/very similarly to how they do in humans. But if they reproduce in vaguely the same way as humans (not asexually, and involving genes and chromosomes and all that) then I don't think they would be “impossible” to exist. If they do reproduce that differently, then;
There's of course a lot of non-genetic causes as well - brain damage, being born premature, micro- and macrocephaly (unusually small and big head respectively), holoprosencephaly, fetal alcohol spectrum disorders, TORCH infections, and a lot more that you could make them have if you want to be more scientifically accurate.
It's also important to remember that a lot of cases of ID don't have an explanation behind them, i.e., no one knows why the person has it. Genetic causes are actually a large minority (like 25%, with majority of that being Down syndrome). Most people who's ID cause is unknown will be on the milder end (kinda by default because most ID people have it mild but still) and usually not have other disabilities, but there's no hard rules. So if you want to not worry about researching causes that would make sense for a bug or a fish, you can say that they were just born like that with no deeper explanation and it would make sense.
What I'd worry about more is to not have the only intellectually disabled character be of an animal/fantasy race that's associated with being mindless and/or child-like. But if all the characters are the same species anyway then it doesn't really apply.
Regardless of what cause (or lack of it) you choose, try to make sure you know what severity their ID is, what symptoms specifically they experience, what can or can't they do, even if the specific diagnosis doesn't ever name-drop in the story. Just be aware that a lot of these conditions have very significant overlap and readers will come up with different conclusions based on their knowledge - someone will assume the character has Down syndrome or autism because these are the only conditions comorbid with ID that they know, someone else will presume 1p36 Deletion syndrome because they have a family member with it - even if you're quite specific about the symptoms. So if you want to represent a specific condition it would be good to drop the actual name, but it's up to you.
I hope I understood the question properly, feel free to send any additional asks/clarifications if I missed the point
mod Sasza
43 notes
·
View notes
Note
Anencephaly, major heart defects, major lung defects, trisomy 13, holoprosencephaly, hydranencephaly, renal agenesis, thanatophoric dysplasia, and triploidy. These aren't "disabilities" ; they are death sentences. You are for forcing women who find out a heart breaking truth to know for months they won't be able to have a baby. You are for forcing an infant into the world only to know torture and agony and pain.
You are a monster, full stop. This is not freedom, this is torture.
Allowing a mother to spend every moment she can with her dying child and also giving her the comfort that she did everything she could rather than the guilt of knowing she had her child killed - that’s what you think is torture?
No family should go through this alone, and there are great resources for perinatal hospice that should be made available to parents.
Also, the number of times doctors have been wrong about a diagnosis or survival chance…
Not to mention new options for correcting issues by performing surgery on a baby in utero to save lives:
Heart surgery for Trisomy 13 and 18, surgery for spina bifida, Renal Anhydramnios Fetal Therapy for renal agenesis, placenta-derived stem cell therapy for spinal bifida, just to name a few
My husband’s parents were told he wouldn’t survive. They were told to abort, and they refused. They planned to say goodbye to him in the hospital after he was born. All the family came. He was born, baptized by his grandpa, and rushed to surgery.
As you probably figured out, he survived (since he is now my husband). He has no lingering effects of the condition that doctors said would kill him, except that he only has one kidney (and doctors now say he has normal kidney function because his single kidney grew to compensate).
Doctors aren’t omniscient. Conditions that were death sentences 20 years ago are now treatable. New surgeries and procedures are constantly being developed.
We’re not going to sentence babies to death because a doctor says they’re going to die. Sorry not sorry.
494 notes
·
View notes
Text
now, i absolutely believe that it is both reasonable and not a dismissal of your concerns for anarchists to tell you they cannot individually provide you a plan for insulin production and distribution, because it depends entirely on your locale--who is in your community, what needs do they have and what skills do they have, what is the actual layout of your locale, what are the optimized transportation options for your available local resources and climate and geography, what local resources are available to create materials i.e. insulin, etc etc etc. plans for resource production & distribution are, fundamentally, not one-size-fits-all in an anarchist framework, and you cannot create those plans without some level of expertise in the relevant topics, which includes expertise in local geography, culture, and demographic makeup.
that said: i do think that, if you are a disabled anarchoprimitivist writing anprim theory through a disabled lens, it is your responsibility to write that theory in a way that encompasses the existence of people who need machines and medications in order to breathe, keep their hearts beating, digest food, and other regulatory body functions, as well as ADLs.
it is not your responsibility to lay out care plans for these people--again, this fundamentally cannot have a one-size-fits-all solution, because you do not have expertise in the needs and resources of every locale on the planet, and you should not be expected to. but your theory needs to be written in a way that explicitly acknowledges the existence of people who need colostomies, use feeding tubes, rely on CPAP machines, need solid organ transplants, people who have spina bifida or holoprosencephaly, et cetera and so on and so forth. this is because theory that does not explicitly deal with the existence of these people is not actually operating in a disability lens.
disability is an extremely broad umbrella. it is, of course, impossible to individually account for every disabled experience; again, that is not the responsibility of anyone writing theory, regardless of the theory in question. but theory written through a disabled lens needs to reckon with the multitude of disabled experiences, foundationally. it isn't a disabled lens if it isn't reckoning with the existence of those unable to breathe without external assistance, and examining what the theory itself looks like if they're centered in its implementation. a disabled lens centers disability, broadly; if an entire massive cohort of disability isn't ever once centered in your theory, even as a thought-experiment aside for a single paragraph, it's not actually engaging with disability as a broad political coalition, it's engaging with your disability or the disabilities of people you care about.
71 notes
·
View notes
Text
As someone who was bullied into carrying a not compatible with life diagnosis (trisomy 13 with holoprosencephaly and cyclopia) to full term (I didn't make it, and my little one only lived eight minutes and suffered horrendously the entire time), fuck all y'all who are supporting these piece of shit politicians and laws.
You're not pro-life. You're pro forced-birth.
I will NEVER forgive myself for going through with things. I will live with my guilt for the rest of my life.
9 notes
·
View notes
Note

Nonbinary person jumpscare
CW for child abandonment, in depth discussion of medical/surgical stuff, and harm to babies
This is Winnipeg, a nonbinary teen (amab and mostly male aligned but very feminine) who was born with several abnormalities- lobar holoprosencephaly and several facial abnormalities, spina bifida, a congenital pulmonary airway malformation, an atrial septal defect, craniosynostosis, and congenital scoliosis.
The primary reason that Winnipeg openly identifies as nonbinary is to take back their personhood after a lifetime of having their body being changed and altered by surgery and let it be a part of them without being all they are. Their pronouns are they/he/xe, and the other kids mostly use they/them for them- to the point that it takes Lottie a HOT minute to learn they even HAVE a separate set of pronouns, let alone two.
Winnipeg was a foundling, left in the Winnipeg General Hospital in Canada- though, this isn’t where he would end up staying, as he ended up moving to the US. The workers at the hospital nicknamed him Winnie, and while they were unsuccessful in tracking down his parents, they assumed he had been abandoned at the hospital because of his numerous medical issues- his eyes were out of place, his lips were split open, his head was misshapen and face too close together, his spine bent with what looked like a large cyst on the bottom, his heartbeat was irregular, and his breathing was poor to the point his split lips and small hands were turning blue.
Over the next several years, Winnipeg underwent numerous surgeries- first heart surgery at only a week old, then craniosynotosis. Their CPAM didn’t need surgery for about six months, but they needed a tracheostomy once their lungs were cleared because of facial abnormalities making it hard for them to breathe. This was taken out after four surgeries to repair their face later on, allowing them to breathe normally. Finally, they underwent surgery for their scoliosis and spina bifida, a spinal fusion in their lower back.
The workers who found him suspect this was why his parents left him- they were likely low income or living in a state of crisis given the scope of Winnipeg’s health problems and the fact that Winnipeg appeared to be a newborn but hadn’t been born in a hospital, and when they saw the state of Winnipeg’s health, knew they would never be able to care for him.
(This is actually what happened- their mother was disabled and in poverty and didn’t think she could take care of a child with high support needs.)
Winnipeg was the one to choose that as their real name as opposed to Winnie.
They’re very chill about almost everything, but sometimes they can get set off by really tiny problems because of how much they have built up. They’re in reality extremely- and justifiably- upset that this is how their life has gone, entirely defined by all of these health issues that even now will never go away. They tend to search for ways to express themself and have control and freedom in their life, often through art. They’re a very artistic person, and they often draw on both their own skin and their friends’. The caretakers are fairly certain that has to be really bad for Ashika’s fragile skin, but they don’t have any real evidence.
Winnipeg is Ashika’s favorite of their group. They can often be found together- Winnipeg sitting in his wheelchair when his pain is bad and Ashika on the floor so she’s not looking down on them, white teddy bear in her lap and one hand, talking to him about anything she can think of while he draws on her free hand.
Fellow “abandoned shortly after being born” person here, I love them already
Him and Ashika interacting is so wholesome, they’re my children now- /j
2 notes
·
View notes
Text
Corpus colossus

It is estimated that at least one in 4,000 individuals has a disorder of the corpus callosum. Impairments in social interaction and communication in individuals having a disorder of the corpus callosum may overlap with autism spectrum disorder behaviors. The corpus callosum acts as a bridge so that input from. Each hemisphere of the brain controls movement and feeling on the opposite side of the body. It acts as a connective pathway that links the left hemisphere and the right hemisphere of the cerebral cortex. Individuals with these disorders have a higher risk of hearing deficits and cardiac abnormalities than individuals with the normal structure. by primary degeneration of the corpus callosum, is a rare complication of chronic alcoholism.1 Although nutritional deficiencies have been suspected. The corpus callosum is a hard, C-shaped structure found in the middle of the brain. Other disorders of the corpus callosum include dysgenesis, in which the corpus callosum is developed in a malformed or incomplete way, and hypoplasia, in which the corpus callosum is thinner than usual. It contains 200 million nerve fibers that pass information back and forth. Children with the most severe brain malformations may have intellectual impairment, seizures, hydrocephalus, and spasticity. The corpus callosum is a structure that connects the right and left sides of the brain. The effects of the disorder range from subtle or mild to severe, depending on associated brain abnormalities. ACC can also be associated with malformations in other parts of the body, such as midline facial defects. Cast and crew from the film Mad Max: Fury Road and celebrities walked the red carpet at Event Cinemas. ACC can occur as an isolated condition or in combination with other cerebral abnormalities, including Arnold-Chiari malformation, Dandy-Walker syndrome, schizencephaly (clefts or deep divisions in brain tissue), and holoprosencephaly (failure of the forebrain to divide into lobes.) Girls may have a gender-specific condition called Aicardi syndrome, which causes severe cognitive impairment and developmental delays, seizures, abnormalities in the vertebra of the spine, and lesions on the retina of the eye. Pictured: Quentin Kenihan Companion (Corpus Colossus). The corpus callosum transfers motor, sensory, and cognitive information between the brain hemispheres. It connects the left and right sides of the brain, allowing for communication between both hemispheres. It is caused by a disruption of brain cell migration during fetal development. The corpus callosum is a thick band of nerve fibers that divides the cerebral cortex lobes into left and right hemispheres. In ACC the corpus callosum is partially or completely absent. Agenesis of the corpus callosum (ACC) is one of several disorders of the corpus callosum, the structure that connects the two hemispheres (left and right) of the brain.
2 notes
·
View notes
Text
Victor McKusick, “Mendelian Inheritance in Man”, 1966, Chromosome #18.
Here I present: Victor McKusick, “Mendelian Inheritance in Man”, 1966, Chromosome #18. Thirty-five (35) traits of chromosome #18 are listed BELOW.
Myopia, high grade, autosomal dominant.
Holoprosencephaly.
Torsion dystonia, adult-onset, focal.
Orthostatic hypotensive disorder of Streeten.
Hepatitis B virus integration site.
Retinoblastoma-binding protein.
Amyloid neuropathy,…

View On WordPress
0 notes
Text
neural tube defects, hydrocephalus, anencephaly with or without herniated neural elements, holoprosencephaly, microcephaly, caudal dysgenesis, hydranencephaly
0 notes
Text
My favorite fact about science is that there is a gene in the human body known as the SHH gene, or the Sonic Hedgehog gene, which sounds like it would be something minor bc they named it after fictional funnyguy, but no, it's believed that variations of the Sonic Hedgehog gene can result in the brain not properly splitting into two hemispheres, causing a disorder known as holoprosencephaly which at worst has a life expectancy of about five hours.
1 note
·
View note
Text
At her 20-week ultrasound appointment, Beaton said her physician discovered the fetus had a rare, severe anomaly -- called alobar holoprosencephaly -- in which the fetus's brain does not develop into two hemispheres as it normally would, and the major structures of the brain remain fused in the middle.
The brain splitting into two hemispheres is a "critical stage in the development" and can impact the development of the nose, mouth and throat, Dr. Katie McHugh, an Indiana OB-GYN and abortion provider, told ABC News. The condition can result in a very painful life and death for the fetus, McHugh said.
0 notes
Text
Trisomy 18 and Microdeletion 18p Mosaicism: A case report and literature review by Chao-Chun ZOU in Journal of Clinical Case Reports Medical Images and Health Sciences

ABSTRACT
The trisomy 18 syndrome is a common chromosomal disorder due to the presence of an extra chromosome 18, either complete, mosaic trisomy or partial trisomy 18q. The mosaic trisomy 18 patients’ phenotype was extremely variable, from the absence of dysmorphic features to complete trisomy 18 syndrome. The phenotype of 18p deletion syndrome is variable and almost all survived. A 2-year-old girl was referred to our hospital due to growth delay. Mild dysmorphy including thin hair, frontal bossing, low set ears, broad-flat nose, nostrils slightly upward, downturned corners of the mouth, dysplasia teeth, small hands and fingers bilaterally was observed. The karyotype of peripheral leukocyte showed 46,XX, psu idic (18)(p11.2)[55]/46,XX, del (18)(p11.2)[45]. We report this case to add to our knowledge of the trisomy 18 and microdeletion 18p mosaicism.
Keywords: Trisomy 18, mosaic;18p microdeletion; Psychomotor retardation; Karyotype
INTRODUCTION
The trisomy 18 syndrome was first reported by Edwards et al in 1960, also known as Edwards syndrome. It is the second most common autosomal chromosomal disorder after trisomy 21(Down’s syndrome)due to the presence of an extra chromosome 18, which has three basic types: complete, mosaic and partial type (Edwards et al., 1960, Cereda and Carey, 2012,Mudaliyar and Mudaliyar, 2017). The syndrome presents a recognizable pattern of major and minor anomalies, significant psychomotor and cognitive disability are associated with high neonatal and infant morbidity and mortality. The estimated overall prevalence of trisomy 18 in live born is approximately 1/6, 000 to 1/8, 000 while the incidence in fetus is much higher, the difference is caused by fetal loss and pregnancy termination after prenatal diagnosis (Cereda and Carey, 2012, Rasmussen et al., 2003). The mosaic trisomy 18 usually means having more than one cell line in the individual, and it occurs in approximately 5 percent in all trisomy 18 patients (Fitas et al., 2013). The phenotypic manifestations are highly variable, from the absence of dysmorphic features to the complete trisomy 18 syndrome (Tucker et al., 2007). Since the clinical outcomes of complete and mosaic trisomy 18 can be different, it is of vital importance to achieve a correct diagnosis because of implications in medical management and genetic counselling. 18p deletion was first described by de Grouchy and colleagues in 1963 and was estimated to occur in approximately 1/50, 000 live born, which results from deletion of a part or full of the short arm of chromosome 18(Turleau, 2008). The mostly reported clinical features include cognitive impairment, congenital heart defects, small stature, minor facial dysmorphy, and skeletal deformities(Turleau, 2008, Xiao et al., 2019, Hasi-Zogaj et al., 2015, Yi et al., 2014)
Typical facial features include hypertelorism, ptosis, strabismus, broad–flat nose, micrognathia, and low-set big ears. Holoprosencephaly may be seen in approximately 10–15% of patients(Turleau, 2008). In addition, speech and language difficulties, pituitary abnormalities, generalized seizures, dystonia, and autoimmune diseases have also been described(Turleau, 2008, Rao et al., 2001, Graziadio et al., 2009, McGoey et al., 2011). However, these non-specific features are easily overlooked clinically. The clinical phenotype severity is related to the size and location of deletion region. In this report, we present a 2-year-old girl of mosaic trisomy 18 and 18p microdeletion with mild psychomotor retardation, cognitive impairment and language developmental disability.
CLINICAL DESCRIPTION
A 2-year-old female second child of non-consanguineous parents was admitted to our hospital due to growth delay. Her mother and father were 34 years old and 38 years old when giving birth to her. She was born at full-term with uncomplicated gestation, her birth weight was 3.35 kg and the length was about 50 cm. No feeding difficulty and complications were referred in the neonatal period. She had a motor retardation of autonomous walking until 22-months old and intelligence disability and language disability. She only knew a few simple words like ”mama“, not ”baba“, and she cannot communicate clearly with others though she was willing to speak to strangers. Gesell Developmental Schedules performed in local hospital indicated mental developmental delay in motor behavior, language behavior, adaptive behavior and personal-social behavior at age of one year and 8 months old. The height of her father, mother and 15
years old sister were 165cm, 161cm, and 155cm, respectively. No similar history was noted in her family.
On physical examination, she had a height of 81.4 cm below -3SD and a weigh of 11.6 kg below -1SD. Mild craniofacial dysmorphy was present, including thin hair, frontal bossing, low set ears, broad-flat nose, nostrils slightly upward, and downturned corners of the mouth while other craniofacial anomalies were not obvious (Fig.1A). Her hands were small especially her fingers, but the fingernails are normal (Fig.1B). Her teeth were dysplasia (Fig.1C). The echocardiography revealed patent foramen ovale (ϕ 2.96 mm) while no murmur was present. The muscle tension was normal and no other organ abnormality was detected in our patient.
Laboratory examinations (urine, liver, kidney, thyroid hormone, GS/MS and blood glucose analyses) were all normal. Insulin-like growth factor-1 was 72.5 ng/ml (normal range, 55-327 ng/ml).
MANAGEMENT AND OUTCOME
Ten months ago, the child was brought to a local hospital with developmental delay, the peripheral leukocyte karyotype was taken and revealed two abnormal cell lines, the result was 46,XX, psu idic (18)(p11.2)[55]/46, XX, del (18)(p11.2)[45] . She was then referred to another hospital to take the whole-exome sequencing demonstrating a deletion at 18p11.32-p11.22 (GRch37/hg19, chr18:158679 9708482del) and a duplication at
18p11.21-q23(GRch37/hg19, chr18:12012132 78005255dup). She was diagnosed mosaic trisomy 18 syndrome.
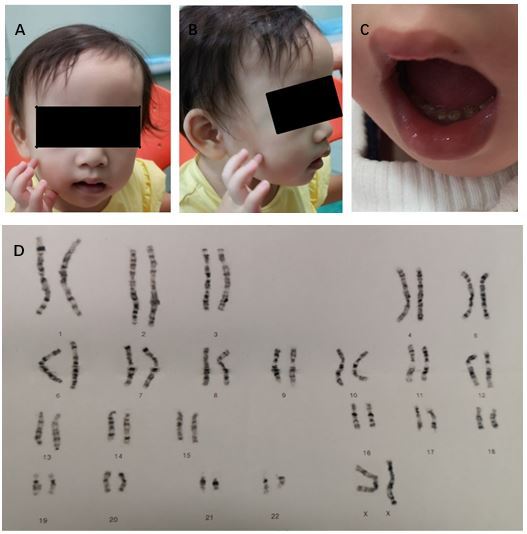
Figure 1: Clinical manifestation and karyotype of our patient. (A and B) thin hair, frontal bossing, low set ears, broad-flat nose, nostrils slightly upward, downturned corners of the mouth, small hands and fingers bilaterally; C) dysplasia teeth; (D) Karyotype shows 46,XX, psu idic (18)(p11.2)[55]/46,XX, del (18)(p11.2)[45]
DISCUSSION
The first reported patients with trisomy 18 syndrome were initially described by Edwards et al and Smith et al in 1960s, while the first case of mosaic trisomy 18 was reported in 1965. Less than 5% portion of patients have mosaicism of trisomy 18, and Banka et al reminded that routine karyotype from lymphocyte culture may not be sufficient to diagnose mosaicism if practitioners suspect a diagnosis of mosaic trisomy 18, karyotype from skin fibroblasts should be considered. Since then over 40 cases of mosaic trisomy 18 have been described, Tucker et al reviewed 33 reported individuals of mosaic trisomy 18 and added 2 more cases in 2007. Their clinical manifestations are extremely variable from complete trisomy 18 syndrome with early death to near totally normal. Some physical features are relatively more common and included brachydactyly, high arched palate, microcephaly, delayed bone age, frequent respiratory infections and otitis media, heart defect, 5th finger clinodactyly, micrognathia, and hypotonia. The most common heart defect is ventricular septal defect in mosaic trisomy 18. Our case has mild craniofacial dysmorphy and patent foramen ovale, and no other physical anomalies were observed.
Trisomy 18 mosaicism usually indicates the existence of more than one cell line in the individual. The peripheral leukocyte karyotype demonstrates pseudodicentric chromosome substituting a normal chromosome 18 in 55 cells and chromosome 18 missing the end of the short arm in 45 cells. The skin fibroblasts karyotype was not taken. Furthermore, there is no correlation between the physical and intellectual findings and the percentage of trisomy 18 cells in either peripheral leukocytes or skin fibroblasts. Besides, there is no correlation between the percentage of trisomic cells in peripheral leukocytes and brain, gonads, or other key organs. The variety of mosaic trisomy 18 may be related to the percentage of trisomic cells in different key organs of the body.
For complete trisomy 18 patients, approximately 50% of infants live longer than one week and about 5-10% of children survive beyond the first year. In overall, trisomy 18 mosaicism patients usually survive longer when compared to complete trisomy 18. This does not mean that all the mosaic trisomy 18 patients have a longer survival, some died a few hours after birth. For normal or mild phenotypical mosaic trisomy 18 cases, some were diagnosed due to recurrent miscarriages or giving birth to a child with trisomy 18 while others may never be identified. 18p deletion syndrome, also called monosomy 18p and De Grouchy syndrome type Ⅰ, which means a deletion of full short arm of chromosome 18 or a microdeletion of the short arm of chromosome 18. Some researches showed that nearly half of patients have breakpoints in the centromeric region and the rest scatter in the short arm, and approximately half of the deletions occur on the maternal chromosome 18 no matter where the breakpoint locations are. Our case’s breakpoint is at the 18p11.32-p11.22. Approximately two thirds of patients’18p deletion are de novo; the rest may be due to a de novo unbalanced translocation or malsegregation of parental chromosome rearrangement or a ring chromosome. The patient’s height and weight is 81.4 cm below -3SD, 11.6 kg below -1SD, respectively. It may be a prodrome of small stature, but her insulin-like growth factor-1 was normal. It also could be contributed to feeding problem. More follow-up work needs to be done to figure it out. Some reported cases show that growth hormone replacement treatment is efficient in growth hormone deficiency patients.
Our case has trisomy 18 and microdeletion 18p mosaicism simultaneously. The possibility of meiotic chromosomal nondisjunction of the ovogonia/spermatocyte was increased because of her parents’ advanced maternal age, some women may have higher a risk for nondisjunction. More possible mechanism may be a de novo unequal recombination occurring in early embryonic mitosis. Some deletions are from the parents, there is no way to figure her mutation mechanism out since we can not get her parents’ consent to analysis. The phenotype of our case combines two syndromes’ typical features, including common psychomotor retardation, cognitive impairment and congenital heart defect, characteristic small stature and language impairment of 18p deletion syndrome. Our case’s uncharacteristic craniofacial features also combine two syndromes.
In a conclusion, mosaic trisomy 18 and 18p deletion syndrome both are chromosomal disorders which has a variety of clinical manifestations. If an individual has untypical phenotypical anomalies and psychomotor and cognitive disability, chromosome disorder should be considered and cytogenic analysis is needed.
Acknowledgements: We thank the patient and his parents for permitting us to use the data.
For more information: https://jmedcasereportsimages.org/about-us/
For more submission : https://jmedcasereportsimages.org/
#Trisomy 18#mosaic#18p microdeletion#Psychomotor retardation#Karyotype#trisomy 18 syndrome#Chao-Chun ZOU#jcrmhs
1 note
·
View note
Last Seen Blogs

fuckyeahpipesofpan
Pipes of Pan

new-york-no-shoes
New-York-No-Shoes

noisypandafire
Untitled
artistcam-mtl
Rodney Gardner Photography

fallstruster
FallsTru2t3r