#Reactive Oxygen Species (ROS)
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
If you dial 1-866-584-6757, you can leave an audio post for your followers.
Text
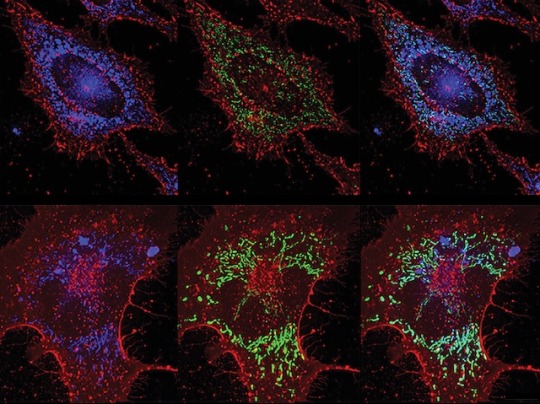
ROS vs Bacteria
Inducing lung lining cells to produce bacteria-killing reactive oxygen species (highly reactive chemicals that can cause oxidative damage) protects against pneumonia without reliance on antibiotics
Read the published research paper here
Image from work by Yongxing Wang and colleagues
Department of Pulmonary Medicine, University of Texas MD Anderson Cancer Center, Houston, TX, USA
Image originally published with a Creative Commons Attribution 4.0 International (CC BY 4.0)
Published in PLOS Pathogens, September 2023
You can also follow BPoD on Instagram, Twitter and Facebook
#science#biomedicine#immunofluorescence#biology#reactive oxygen species#ROS#sci art#pulmonary#lungs#pneumonia#antibiotics#antibiotic resistance
10 notes
·
View notes
Text
The Role of Mitochondria in Autism Spectrum Disorder

Introduction
Autism Spectrum Disorder (ASD) is a neurodevelopmental condition defined by difficulties in communication, social interaction, and the presence of repetitive behaviors. While ASD's exact origins remain complex and multifaceted, growing research highlights mitochondrial dysfunction as a key biological contributor. Mitochondria are vital organelles responsible for generating cellular energy and maintaining homeostasis, particularly in energy-demanding organs like the brain. Impairments in mitochondrial function can significantly disrupt neural development and have been increasingly observed in individuals with ASD. This article delves into the role of mitochondria in ASD, exploring evidence of mitochondrial abnormalities and their implications for understanding and treating this condition.
Mitochondrial Function and Brain Development
Mitochondria produce adenosine triphosphate (ATP), the energy currency of cells, through a process known as oxidative phosphorylation. This energy production is essential for many brain processes including neurotransmission, synaptic plasticity, and cellular repair. Besides energy generation, mitochondria are also involved in regulating calcium levels, producing reactive oxygen species (ROS), and controlling apoptosis (programmed cell death). All these functions are particularly important during early brain development when neurons are rapidly forming connections and networks.
Mitochondrial Dysfunction in Autism
Abnormal Biochemical Profiles Numerous studies have detected elevated levels of lactate, pyruvate, and alanine in the blood and cerebrospinal fluid of individuals with ASD. These findings suggest a disruption in mitochondrial energy metabolism. Abnormal lactate-to-pyruvate ratios, for example, point to oxidative phosphorylation inefficiencies, implying that mitochondria in ASD-affected individuals are not functioning optimally.
Increased Oxidative Stress Mitochondria are both producers and targets of ROS, and when not properly regulated, ROS can damage DNA, proteins, and lipids. In individuals with ASD, elevated markers of oxidative stress and reduced levels of antioxidants such as glutathione have been reported. This imbalance can contribute to neural inflammation and impair neurodevelopment, possibly exacerbating core ASD symptoms.
Genetic Abnormalities in Mitochondrial DNA Some individuals with ASD exhibit mutations or deletions in mitochondrial DNA (mtDNA). Since mtDNA is crucial for the normal function of the electron transport chain—essential for ATP production—such mutations can compromise cellular energy availability. Furthermore, mitochondrial diseases, which often involve mtDNA mutations, frequently present with neurodevelopmental symptoms overlapping with those of ASD.
Mitochondrial Dynamics and Quality Control Mitochondrial health depends on processes such as fission, fusion, and mitophagy (the removal of damaged mitochondria). In ASD, studies have observed altered expressions of genes involved in these dynamic processes. Imbalances in mitochondrial fission and fusion can lead to dysfunctional mitochondria accumulating in neurons, impairing their function and survival.
Impact on Synaptic Function Efficient synaptic transmission relies heavily on mitochondrial energy. Mitochondria located at synapses help regulate calcium signaling and provide the necessary ATP for neurotransmitter release. Mitochondrial dysfunction may therefore contribute to the synaptic abnormalities frequently observed in ASD, including disruptions in excitatory/inhibitory balance, which are believed to underpin many behavioral features of the disorder.
Therapeutic Approaches Targeting Mitochondrial Dysfunction
Understanding mitochondrial involvement in ASD opens the door to potential targeted therapies. Several interventions are currently being explored:
Antioxidant Therapy: Compounds such as coenzyme Q10, alpha-lipoic acid, and N-acetylcysteine have been investigated for their ability to reduce oxidative stress and improve mitochondrial function.
Mitochondrial Cofactor Supplementation: Nutrients like L-carnitine, B-vitamins, and creatine that support mitochondrial metabolism are being studied for their efficacy in alleviating certain ASD symptoms.
Dietary Strategies: Diets such as the ketogenic diet, which alters energy metabolism to rely more on ketone bodies, have shown potential in improving mitochondrial function and behavior in some individuals with ASD.
While these approaches offer promise, it is essential that treatments are personalized and medically supervised, as mitochondrial involvement varies widely among individuals with ASD.
Conclusion
The emerging link between mitochondrial dysfunction and Autism Spectrum Disorder provides a valuable lens through which to understand this complex condition. By affecting energy production, synaptic regulation, and oxidative balance, mitochondria may play a pivotal role in ASD pathogenesis. Further research is needed to refine our understanding and to develop effective, targeted treatments. Nonetheless, recognizing the role of mitochondria enhances our broader understanding of neurodevelopmental disorders and holds promise for future therapeutic innovations that may improve outcomes for individuals on the autism spectrum.
on as a key biological contributor. Mitochondria are vital organelles responsible for generating cellular energy and maintaining homeostasis, particularly in energy-demanding organs like the brain. Impairments in mitochondrial function can significantly disrupt neural development and have been increasingly observed in individuals with ASD. This article delves into the role of mitochondria in ASD, exploring evidence of mitochondrial abnormalities and their implications for understanding and treating this condition.
Mitochondrial Function and Brain Development
Mitochondria produce adenosine triphosphate (ATP), the energy currency of cells, through a process known as oxidative phosphorylation. This energy production is essential for many brain processes including neurotransmission, synaptic plasticity, and cellular repair. Besides energy generation, mitochondria are also involved in regulating calcium levels, producing reactive oxygen species (ROS), and controlling apoptosis (programmed cell death). All these functions are particularly important during early brain development when neurons are rapidly forming connections and networks.
Mitochondrial Dysfunction in Autism
Abnormal Biochemical Profiles Numerous studies have detected elevated levels of lactate, pyruvate, and alanine in the blood and cerebrospinal fluid of individuals with ASD. These findings suggest a disruption in mitochondrial energy metabolism. Abnormal lactate-to-pyruvate ratios, for example, point to oxidative phosphorylation inefficiencies, implying that mitochondria in ASD-affected individuals are not functioning optimally.
Increased Oxidative Stress Mitochondria are both producers and targets of ROS, and when not properly regulated, ROS can damage DNA, proteins, and lipids. In individuals with ASD, elevated markers of oxidative stress and reduced levels of antioxidants such as glutathione have been reported. This imbalance can contribute to neural inflammation and impair neurodevelopment, possibly exacerbating core ASD symptoms.
Genetic Abnormalities in Mitochondrial DNA Some individuals with ASD exhibit mutations or deletions in mitochondrial DNA (mtDNA). Since mtDNA is crucial for the normal function of the electron transport chain—essential for ATP production—such mutations can compromise cellular energy availability. Furthermore, mitochondrial diseases, which often involve mtDNA mutations, frequently present with neurodevelopmental symptoms overlapping with those of ASD.
Mitochondrial Dynamics and Quality Control Mitochondrial health depends on processes such as fission, fusion, and mitophagy (the removal of damaged mitochondria). In ASD, studies have observed altered expressions of genes involved in these dynamic processes. Imbalances in mitochondrial fission and fusion can lead to dysfunctional mitochondria accumulating in neurons, impairing their function and survival.
Impact on Synaptic Function Efficient synaptic transmission relies heavily on mitochondrial energy. Mitochondria located at synapses help regulate calcium signaling and provide the necessary ATP for neurotransmitter release. Mitochondrial dysfunction may therefore contribute to the synaptic abnormalities frequently observed in ASD, including disruptions in excitatory/inhibitory balance, which are believed to underpin many behavioral features of the disorder.
Therapeutic Approaches Targeting Mitochondrial Dysfunction
Understanding mitochondrial involvement in ASD opens the door to potential targeted therapies. Several interventions are currently being explored:
Antioxidant Therapy: Compounds such as coenzyme Q10, alpha-lipoic acid, and N-acetylcysteine have been investigated for their ability to reduce oxidative stress and improve mitochondrial function.
Mitochondrial Cofactor Supplementation: Nutrients like L-carnitine, B-vitamins, and creatine that support mitochondrial metabolism are being studied for their efficacy in alleviating certain ASD symptoms.
Dietary Strategies: Diets such as the ketogenic diet, which alters energy metabolism to rely more on ketone bodies, have shown potential in improving mitochondrial function and behavior in some individuals with ASD.
While these approaches offer promise, it is essential that treatments are personalized and medically supervised, as mitochondrial involvement varies widely among individuals with ASD.
Conclusion
The emerging link between mitochondrial dysfunction and Autism Spectrum Disorder provides a valuable lens through which to understand this complex condition. By affecting energy production, synaptic regulation, and oxidative balance, mitochondria may play a pivotal role in ASD pathogenesis. Further research is needed to refine our understanding and to develop effective, targeted treatments. Nonetheless, recognizing the role of mitochondria enhances our broader understanding of neurodevelopmental disorders and holds promise for future therapeutic innovations that may improve outcomes for individuals on the autism spectrum.
#Autism Spectrum Disorder#Mitochondrial dysfunction#Mitochondria and autism#Neurodevelopmental disorders#Mitochondrial DNA mutations#Oxidative stress in autism#ATP production#Synaptic dysfunction in ASD#Mitochondrial energy metabolism#Reactive oxygen species (ROS)#Mitochondrial cofactor therapy#Antioxidant therapy for autism#Ketogenic diet and autism#Calcium signaling in neurons#Lactate-to-pyruvate ratio#Autism mitochondrial biomarkers#Mitochondrial fission and fusion#Early brain development#Mitochondrial therapeutic strategies#Neuroinflammation in autism
0 notes
Text
youtube
#WO₃₋ₓ@Ferrocene-Folic Acid#photothermal therapy#chemodynamic therapy#reactive oxygen species#Fenton reaction#cancer nanomedicine#targeted drug delivery#tumor microenvironment#immunogenic cell death#nanoplatform#folate receptor targeting#oxidative stress#tumor ablation#ferrocenyl compounds#nanoparticle-mediated therapy#oxygen-deficient tungsten oxide#cancer immunotherapy#ROS generation#synergistic therapy#advanced oncology treatments.#Youtube
1 note
·
View note
Text
The role of antioxidants in periodontal health | ICPA Health Products Ltd.

Periodontal disease is a persistent inflammatory condition that impacts the tissues encircling our teeth. This condition arises from an imbalance between oral biofilms and the body’s immune response, potentially leading to the loss of tooth-supporting tissues. When this inflammation is confined to the protective periodontium, it’s termed gingivitis. However, when it extends to the periodontal supporting structures, it’s recognized as periodontitis. This prevalent oral disease, affecting 10%-15% of adults, is primarily driven by bacterial plaque microorganisms. Additionally, factors such as systemic health, oral hygiene, age, gender, and smoking play significant roles in its development.
The double-edged sword of Reactive Oxygen Species (ROS)
Reactive oxygen species (ROS) serve as crucial players in our body’s defense against invading pathogens. These molecules have antimicrobial properties that help combat infections in the oral cavity. However, ROS can be a “double-edged sword” since an excessive presence of these molecules can become cytotoxic to our own cells. ROS plays pivotal roles in cell signaling, gene regulation, and antimicrobial defense. An overabundance of ROS, coupled with inadequate antioxidant capacity, can result in oxidative stress within the affected periodontal tissues. This, in turn, leads to pathological changes and the destruction of host tissues, ultimately culminating in the loss of teeth as their supporting structures degrade. Inside cells, ROS can inflict damage on biomolecules and cell membranes, further exacerbating the situation.
The link between oxidative stress and periodontal disease
Emerging research has elucidated the connection between oxidative stress and periodontal disease. In the early stages of periodontal disease, especially in the case of periodontitis, a prominent oxidative process unfolds, characterized by elevated levels of reactive oxygen and nitrogen species (ROS and RNS). This oxidative onslaught can upset the balance of the body’s response, triggering changes in biomolecules, especially lipids, proteins, and nucleic acids, ultimately leading to damage to periodontal tissues.
The role of antioxidants
To counteract the deleterious effects of excessive free radicals generated during oxidative stress, our bodies possess an antioxidant defense system. Antioxidants can inhibit and reduce the damage caused by these harmful molecules. These natural antioxidants are found in various sources, including foods, teas, vitamins, minerals, and more. They are also used in auxiliary treatments for conditions such as cardiovascular diseases, pulmonary diseases, aging, and atherosclerosis, all of which share physiological links with periodontal diseases. This suggests that the application of antioxidants might also yield benefits in managing periodontal health.
Promising results
Recent scientific inquiry supports the idea that antioxidants can play a pivotal role in the treatment of periodontitis. A meta-analysis comprising fifteen clinical trials demonstrated uniformly positive outcomes associated with antioxidant supplementation during periodontitis treatment. These findings offer hope and promise for those seeking alternative therapies to complement traditional periodontal treatments.
Conclusion
As we delve deeper into the intricacies of periodontal disease, the role of oxidative stress and antioxidants emerges as a significant area of interest. Oxidative stress appears to be a key contributor to the development and progression of periodontal
diseases. Antioxidants, with their ability to neutralize harmful free radicals, hold promise as adjunct therapies in managing and mitigating the effects of periodontal disease. With ongoing research in this field, we are one step closer to a more comprehensive understanding of periodontal health and the potential for novel treatments that can preserve our smiles for years to come.
#Periodontal disease#Reactive oxygen species (ROS)#periodontal tissues#oral disease#oxidative stress and periodontal disease.#What is oxidative stress?#dental care#application of antioxidants#treatment of periodontitis#periodontal health
0 notes
Text

Horses are among the world’s most elite athletes: When galloping, they can consume twice as much oxygen per kilogram as the fittest humans. All that oxygen supercharges horses’ cells’ energy-producing compartments as they crank out ATP, the chemical needed to power their impressive muscles. But making so much cellular fuel so quickly comes with a catch: the manufacture of pernicious byproduct molecules called reactive oxygen species (ROS), which can wreak havoc in cells.
How horses dealt with this biological trade-off and evolved into premier endurance athletes has long intrigued biologists. Researchers report today in Science that they have uncovered a big part of it, identifying a key mutation that lets horses safely produce so much ATP. The trait helped pave the way for horses to go from dog-size critters millions of years ago to the high-endurance athletes we know today.
The study’s detailed molecular work makes it “exceptional,” says José Calbet, an expert on the cellular responses to exercise at the University of Las Palmas de Gran Canaria who wasn’t involved with the study.
The mutation in question occurs in the gene that encodes a protein called KEAP1, which acts as a biochemical bouncer, binding to a different protein called NRF2 to prevent it from entering the cell’s nucleus, where it would otherwise activate stress-response genes that help blunt cell damage.
But ROS can help NRF2 sneak in by causing KEAP1 to release its bind on the protein, allowing it to enter the nucleus and trigger the cell’s stress-response genes.
Johns Hopkins University ophthalmologist and clinician scientist Elia Duh, a senior author of the new study, didn’t set out to study horses. Initially, Duh was interested in the KEAP1-NRF2 system because its role in activating stress-response genes makes it a tempting target for treating inflammation—and aging-related conditions, such as blinding retinal diseases, irritable bowel syndrome, and neurodegeneration.
Duh wondered whether any insights could be gleaned from studying the evolution of these proteins in different animals. So, he teamed up with Gianni Castiglione, an evolutionary biologist and biochemist at Vanderbilt University. Together, they scanned hundreds of vertebrate genomes looking for notable mutations to the gene for KEAP1.
The team’s genomic work revealed birds had almost completely lost the gene, presumably an adaptation to the extreme demands of flight. When they looked in horses, researchers noticed what initially appeared to be a DNA sequence that encoded an unusually short—and therefore presumably nonfunctional—version of the KEAP1 protein. But when Duh’s and Castiglione’s team grew horse cells in culture, it discovered the protein was very much there and working. “Naturally, I was worried I was doing something wrong,” Castiglione says. “Then one day, a light bulb went off.”
As it turns out, the computer algorithm scientists had used to scan the horse genome had made a mistake. The algorithm had spotted a specific kind of mutation in the part of the KEAP1 gene that changed the messenger RNA from CGA—which codes for the amino acid arginine—to UGA, which is what’s known as a “stop codon.”
Normally, the cellular machinery interprets UGA as a sign to stop translating the RNA into a protein. But instead, the horses’ genetic machinery recodes the stop codon into a different amino acid, cysteine, causing it to ignore that order. This phenomenon, known as a stop codon read-through, is common among viruses but rare in multicellular organisms.
“The identification of this evolutionarily significant UGA recoding event represents a potentially seminal finding, offering a model for uncovering other yet-unidentified cases of stop codon read-through,” says Hozumi Motohashi, a biologist at Tohoku University who has studied KEAP1 and NRF2.
That the replacement is a cysteine is particularly notable, Castiglione says. KEAP1 senses cellular stress through its cysteines, which contain sulfur atoms whose reactions with ROS, induce the chemical changes that cause KEAP1 to let go of NRF2. The mutation the researchers had identified adds another place on KEAP1 for ROS to interact, which makes the protein more sensitive to stress—and lets horse cells respond much faster to the cellular stress of intense exercise. “It does make complete sense [that] by introducing another cysteine, another sulfur, you would have heightened sensitivity,” Castiglione says.
What’s more, this tweaking of KEAP1 is a “[key] genetic component to the puzzle of the evolution of horses,” Duh says. “Once they figured out how to run, they could occupy all kinds of ecological niches,” Castiglione adds.
The finding could also point the way toward new kinds of drugs to treat diseases by targeting the specific parts of the KEAP1 protein that help horses hoof it. “By looking at what evolution has figured out, we know this is a viable strategy,” Castiglione says.
Source
834 notes
·
View notes
Text
Also preserved in our archive
Listen at the first link!
The GIST
Recent studies suggest that a hypermetabolic state that damages the mitochondria results in a hypometabolic state in chronic fatigue syndrome (ME/CFS), long COVID, and fibromyalgia (FM). They also suggest that something in the blood, serum, or plasma is damaging the mitochondria in these diseases.
We’re not done with the mitochondria, though – far from it! Now we look at a bevy of recent long-COVID mitochondrial studies suggesting that mitochondrial dysfunction affects more than energy production and which illuminate what may have gone wrong in the mitochondria.
Muscle biopsies of 120 long-COVID patients who had ended up in the ICU found that a year later their muscles had higher levels of immune cells involved in tissue repair and reduced activity of the 2nd and fourth mitochondrial complexes. The authors concluded that there was “aberrant repair and altered mitochondrial activity in skeletal muscle.”
They couldn’t explain how a respiratory illness affected the muscles but a subsequent study did. A hamster model found that the coronavirus suppressed the genes associated with the muscle fibers, protein production, both sides of the mitochondrial energy production process (Krebs cycle and electron transport chain), and fat breakdown.
As it was doing that, it unleashed a barrage of inflammatory factors (IFN-α, IFN-γ, and TNF-α) which triggered a shift from relying mostly on aerobic energy production to the less effective process of anaerobic energy production (glycolysis).
The authors concluded that using treatments “that can boost mitochondrial functions, enhance protein synthesis, and inhibit protein degradation” may be useful for treating muscle fatigue in long COVID.
Next, a muscle study assessing “maximal fatty acid oxidation (MFO)” (i.e. energy produced by the breakdown of fats during exercise) found significantly reduced levels of fatty acid oxidation in long COVID and a “premature shift” from relying on fats to carbohydrates to powering their cells.
This was important because the body prefers to burn fats during exercise and because fats play key roles in both parts of the mitochondrial energy production process. The finding wasn’t so surprising, though. Problems with carnitine – which transports fatty acids into the mitochondria – have popped up in both long COVID and ME/CFS – suggesting that the fatty acids that power the mitochondria during exercise may not be getting into them.
A review paper asserted that increased free radical production (reactive oxygen species (ROS)) by the mitochondria both pushes the cell into a state of anaerobic energy production but also pushes the immune system to activate the inflammatory or innate immune response and away from the adaptive immune response that targets pathogens. This benefits the viruses by providing the substrates they need to grow and allows them to escape from the immune system.
Several researchers, including Avindra Nath, believe that the immune system tries to compensate for the impaired adaptive immune defense by ramping up the innate immune response. Nath believes this shift plays a central role in ME/CFS.
They proposed that treatments to boost mitochondrial functioning and reduce the production of mitochondrial reactive oxygen species (ROS) (free radicals) will be beneficial.
Lastly, a review asserted that the predominant view of the mitochondria as the main energy producers of the cell is misguided and incomplete. Harkening back to Naviaux’s characterization of the mitochondria as the primary threat-sensing part of the cell, the authors believe the mitochondria regulate the “physiological processes at the level of the cell, organ and organism”; i.e. the mitochondrial problems affect much more than low energy levels and fatigue.
A blog on red light/infrared light therapy – which could both boost mitochondrial health and antioxidant defenses – is coming up.
Full text at either link! There's a lot more than the gist
#long covid#covid is airborne#pandemic#mask up#covid#wear a mask#public health#covid 19#wear a respirator#still coviding#coronavirus#sars cov 2
92 notes
·
View notes
Text
Now this takes me back to a 20 year old hyperfixation on herbs and plants with medicinal benefits. I did not expect to find this in my search.
70 notes
·
View notes
Text
If we are going to reverse the mental illness epidemic in America, we need research on mild, long-term aluminum chelation protocols funding by NIH: deferoxamine (aka desferoxamine), intranasal insulin, silicic acid, cilantro, and glutathione enhancers. And we need to stop inject aluminum into ourselves. Join me in calling on Dr. Jay Bhattacharya to have his Program Officers write calls for proposals for this urgent area of clinical research to reverse iatrogenic disease caused by injected aluminum.
Aluminum (Al) is a highly abundant metal with no known physiological role in the human body. Despite its frequent use in industrial, pharmaceutical, and consumer products, aluminum is a known neurotoxin. Chronic exposure has been implicated in the pathogenesis of multiple neuropsychiatric and neurodegenerative disorders. While aluminum toxicity was once dismissed as irrelevant due to assumed poor absorption and rapid clearance, a growing body of research shows that aluminum bioaccumulates in vulnerable tissues (Tomljenovic et al., 2013) , particularly the brain, and may be a major avoidable risk factor in the development of Alzheimer’s disease (AD; Armstrong et al., 2019), autism spectrum disorder (ASD; Boretti, 2021; Roe, 2022), dialysis encephalopathy (Alfrey et al., 1978; Mach et al. 1988), and other cognitive and behavioral disorders such as acute aluminum intoxication, which requires chelation therapy.
Aluminum exerts its neurotoxicity through several well-documented mechanisms:
Oxidative Stress: Aluminum promotes the generation of reactive oxygen species (ROS), lipid peroxidation, and oxidative DNA damage, leading to mitochondrial dysfunction and neuronal death.
Mitochondrial Disruption: Aluminum impairs mitochondrial energy metabolism and promotes apoptotic pathways, contributing to neurodegeneration.
Metal Mimicry and Ion Disruption: By mimicking essential ions such as Ca²⁺ and Fe³⁺, aluminum disrupts neuronal signaling, membrane potentials, and ferritin iron storage systems.
Epigenetic Modulation: Aluminum alters DNA methylation patterns, histone acetylation, and the expression of microRNAs (e.g., miR-29a/b, miR-124; Aschne et al., 2024), contributing to dysregulated amyloid precursor protein (APP) and tau pathology in Alzheimer’s disease (Kandimalla et al., 2016;Huat et al., 2019).
Autophagy Dysfunction: Aluminum impairs neuronal autophagy, compromising the clearance of misfolded proteins such as β-amyloid and hyperphosphorylated tau (Sanajou et al., 2023; Makhdoomi et al., 2023).
8 notes
·
View notes
Text
you know taking a radiation biology class has made me like my url a lot more. niche reference to ro's old band name now it's like haha i am killing you with low let particles that penetrate your entire body. i am generating reactive oxygen species and damaging your dna. you will develop cancer in about 30 years
#unless it's like an acute high dose i guess. then you'll get radiation sickness. and die#but slowly developing cancer is funnier to me#don't take that out of context btw
14 notes
·
View notes
Text
A team of scientists has developed a new solution for the treatment of rheumatoid arthritis (RA). The work has been published in Nature Nanotechnology. RA is a chronic disease that, unfortunately, has no cure. The disease triggers a mix of troublesome symptoms like inflamed joints, harmful cytokines, and immune system imbalances, which work together to create a relentless cycle of worsening symptoms. While targeting some of these factors can provide short-term relief, others remain unresolved, leading to a frustrating cycle of remission and flare-ups. One of the major hurdles in RA treatment is the inability to restore the immune system to its healthy state. This leaves the body unable to control the continuous production of harmful substances like reactive oxygen species (ROS) and inflammatory cytokines, leading to persistent inflammation and discomfort. In essence, the ideal treatment for RA should not only provide immediate relief from inflammation and symptoms but also address the root cause by restoring the immune system to its normal, balanced state.
Continue Reading
142 notes
·
View notes
Text
Effect of Pollution on Mitochondria: Mechanisms and Implications for Human Health

Introduction
Rapid industrialization and urban development have significantly increased environmental pollution levels, particularly in urban areas. Air pollution is a complex mixture of gases (like ozone, nitrogen dioxide, carbon monoxide) and particulate matter, with PM2.5 being of particular concern due to its ability to penetrate deeply into the alveoli and even enter the bloodstream.
Mitochondria are critical for ATP synthesis, calcium regulation, and programmed cell death (apoptosis). They also regulate redox signaling and cellular metabolism. Given their central role in maintaining cellular integrity and their sensitivity to oxidative stress, mitochondria are prime targets for damage induced by air pollutants. Mitochondrial dysfunction caused by pollution contributes to a wide range of diseases, including neurodegenerative disorders, cardiovascular diseases, and metabolic syndromes.
Mechanisms of Mitochondrial Damage by Pollution
1. Oxidative Stress and ROS Generation
The most prominent mechanism through which pollution affects mitochondria is oxidative stress. PM2.5 and other pollutants contain transition metals and organic compounds that catalyze the formation of reactive oxygen species (ROS). When ROS production exceeds the cell’s antioxidant capacity, it leads to oxidative damage of mitochondrial lipids, proteins, and DNA.
Pollutant-induced oxidative stress disrupts the electron transport chain (ETC), particularly Complex I and III, which further elevates ROS production. This cycle of ROS-induced ROS release exacerbates mitochondrial damage, leading to a decline in membrane potential and ATP production.
2. Mitochondrial Membrane Potential Disruption
The mitochondrial membrane potential (Δψm) is essential for ATP generation through oxidative phosphorylation. Exposure to air pollutants like diesel exhaust particles and PM2.5 causes depolarization of Δψm. This loss of potential impairs ATP synthesis, alters calcium homeostasis, and activates mitochondrial permeability transition pores (mPTP), promoting cell death.
Electron microscopy studies have shown that pollutant-exposed cells exhibit swollen mitochondria, disrupted cristae, and fragmented networks—hallmarks of severe mitochondrial dysfunction.
3. Mitochondrial DNA (mtDNA) Damage
Unlike nuclear DNA, mtDNA lacks protective histones and has limited repair mechanisms, making it highly vulnerable to ROS. PM2.5 and ozone exposure have been shown to cause strand breaks, deletions, and mutations in mtDNA. This impairs the expression of key mitochondrial proteins, further disrupting the ETC and leading to chronic energy deficits.
Mitochondrial DNA copy number has also been used as a biomarker for oxidative stress in epidemiological studies. Decreased mtDNA content correlates with pollution exposure and poor health outcomes in both children and adults.
4. Induction of Apoptosis and Necrosis
Mitochondria regulate both intrinsic apoptotic and necrotic cell death pathways. Air pollutants trigger apoptosis by promoting the release of pro-apoptotic factors such as cytochrome c, apoptosis-inducing factor (AIF), and Smac/DIABLO into the cytosol. These factors activate caspases and lead to programmed cell death.
Additionally, high levels of ROS and persistent mitochondrial dysfunction can shift the balance toward necrosis—an uncontrolled form of cell death characterized by inflammation and tissue damage.
5. Impaired Mitophagy and Biogenesis
Mitophagy is the selective degradation of damaged mitochondria. Air pollution can inhibit mitophagy by altering signaling pathways involving PINK1 and Parkin, leading to the accumulation of dysfunctional mitochondria. Conversely, some pollutants may overstimulate mitophagy, causing loss of healthy mitochondria.
Furthermore, air pollutants downregulate genes associated with mitochondrial biogenesis, such as PGC-1α, NRF1, and TFAM. This leads to reduced mitochondrial number and impaired cellular resilience to oxidative stress.
Health Implications of Pollution-Induced Mitochondrial Dysfunction
1. Cardiovascular Diseases
Endothelial cells, which line blood vessels, rely on functional mitochondria to regulate vascular tone and integrity. Pollution-induced mitochondrial damage in these cells leads to endothelial dysfunction, reduced nitric oxide production, and increased vascular inflammation—key precursors to atherosclerosis, hypertension, and myocardial infarction.
2. Respiratory Conditions
Inhaled pollutants directly affect lung epithelial and alveolar macrophage mitochondria. Damage to these cells can result in chronic obstructive pulmonary disease (COPD), asthma, and reduced lung function. Mitochondrial dysfunction increases susceptibility to infections and reduces the lung’s ability to clear particulate matter.
3. Neurological Disorders
The brain is particularly sensitive to mitochondrial impairment due to its high energy demand. Pollutants like ultrafine particles can cross the blood-brain barrier and accumulate in neural tissue. Studies show that exposure to PM2.5 induces mitochondrial fragmentation, synaptic dysfunction, and neuroinflammation. These changes are associated with increased risks for Alzheimer’s disease, Parkinson’s disease, and cognitive decline.
4. Metabolic Disorders and Diabetes
Mitochondria are central to metabolic homeostasis. Pollutants disrupt mitochondrial function in adipose tissue, liver, and muscle, leading to insulin resistance and impaired glucose metabolism. Epidemiological studies have linked air pollution exposure to increased incidence of type 2 diabetes and obesity.
5. Reproductive and Developmental Effects
Pollution-induced mitochondrial dysfunction can affect gametogenesis, embryo development, and placental function. Prenatal exposure to air pollution has been associated with low birth weight, preterm birth, and developmental delays—possibly due to mitochondrial damage in placental and fetal tissues.
Therapeutic and Preventive Strategies
1. Antioxidant Supplementation
Antioxidants such as vitamin C, vitamin E, Coenzyme Q10, and N-acetylcysteine (NAC) have shown promise in mitigating ROS-induced mitochondrial damage. Mitochondria-targeted antioxidants like MitoQ and SkQ1 are being explored for their ability to penetrate mitochondrial membranes and neutralize ROS at the source.
2. Lifestyle Interventions
Regular physical activity, a diet rich in antioxidants, and stress management can enhance mitochondrial resilience. Avoiding high-pollution areas and using air purifiers indoors can reduce exposure levels, especially in vulnerable populations.
3. Pharmacological Approaches
New therapies targeting mitochondrial biogenesis, dynamics, and repair mechanisms are under investigation. Drugs modulating PGC-1α activity or enhancing mitophagy may offer therapeutic benefit against pollution-induced mitochondrial dysfunction.
Future Research Directions
More research is needed to:
Clarify dose-response relationships between different pollutants and mitochondrial damage.
Investigate the combined effects of multiple pollutants.
Develop non-invasive biomarkers of mitochondrial dysfunction.
Identify genetic or epigenetic factors that influence individual susceptibility.
Explore targeted therapies to prevent or reverse mitochondrial impairment.
Longitudinal and population-based studies will be key in establishing causal links between pollution, mitochondrial dysfunction, and disease progression.
Conclusion
Mitochondria are critical targets of pollution-induced cellular damage. The mechanisms—including oxidative stress, mtDNA damage, impaired mitophagy, and disrupted bioenergetics—converge to impair cellular function and promote disease. As air pollution levels remain a pressing global concern, understanding and addressing mitochondrial responses to environmental toxins is essential for public health. Preventive measures and therapeutic strategies focused on mitochondrial health could play a crucial role in reducing the disease burden associated with pollution.
#Mitochondrial dysfunction#Air pollution and mitochondria#PM2.5 oxidative stress#Pollution-induced ROS#Mitochondrial membrane potential#Mitochondrial DNA damage#Environmental pollutants and health#Mitochondria and air pollutants#Mitophagy impairment#Reactive oxygen species (ROS)#Fine particulate matter effects#Mitochondrial biogenesis inhibition#Pollution-related metabolic disorders#Mitochondrial-targeted antioxidants#Air pollution neurotoxicity#Mitochondria and cardiovascular health#Pollution and mitochondrial diseases#Mitochondrial response to environmental stress#Airborne particles and cell damage
0 notes
Text
youtube
#Photodynamic therapy#aggregation-induced emission#AIE-PSs#photosensitizers#reactive oxygen species#tumor microenvironment#cancer therapy#ROS generation#non-invasive treatment#tumor targeting#cancer research#precision medicine#light activation#photostability#intracellular aggregation#aggregation-enhanced photosensitizers#oncology advancements#tumor selectivity#biophotonics#therapeutic precision.#Youtube
0 notes
Text
Alcohol consumption is a significant risk factor for various cancers, including those of the mouth, throat, esophagus, liver, colon, rectum, and breast. When alcohol is metabolized in the body, it converts to acetaldehyde, a toxic compound that can damage DNA and hinder its repair mechanisms. This DNA damage can lead to uncontrolled cell growth, a hallmark of cancer development.

Additionally, alcohol can generate reactive oxygen species (ROS), leading to oxidative stress and further DNA damage. It also impairs the body's ability to absorb essential nutrients like vitamins A, C, D, E, and folate, which play protective roles against cancer. Moreover, alcohol increases estrogen levels, potentially elevating the risk of breast cancer. Given these mechanisms, it's crucial to be aware of the cancer risks associated with alcohol consumption and consider moderating intake to reduce potential harm.
6 notes
·
View notes
Text

Garlic - helps with blood sugar regulation, antioxidant with anti-inflamatory properties, and assists in lowering blood pressure.
Honey - antioxidant flavonoids and phenolic acids help neutralize reactive oxygen species (ROS) in your body which can build up in cells and cause damage, reduces inflammation, improves blood sugar regulation, improves cholesterol and triglyceride levels, and prevent heart disease.
Chicken - zinc, iron, B vitamins, protein, reduces blood pressure, lowers risk of heart disease
2 notes
·
View notes
Note
No, no, please, go on, S. Aureus has my attention and I wish to know more...
anon you have my eternal gratitude for your continued interest.... WELL what i cut myself off on talking about was the virulence factors! aka what s aureus Has That Makes It Toxic. well first off it can release a bunchhh of enzymes that degrade the environment around.. like it Literally has a desoxyribonuclease to break down DNA! (little shorthand--anything that ends in -ase is usually an enzyme, aka it Breaks Down Something). it can also secrete a bunch of toxins like! superantigens!
so an antigen is what an immune cell recognizes to go Oh this thing is a stranger to our body! we have to destroy it! and those interactions are Specific like different lymphocytes have different Specificities in the antigens they can recognize. a SUPERantigen is one that bypasses those specificities and just activates a MASS amount of lymphocytes. and sure immune cells are there to protect us but there's a balance to maintain! when there's a mass reaction like that it overbalances into toxic shock syndrome (yk .... what they scare you with so you don't keep your tampon in too long). basically your whole body goes into crisis mode.
they can ALSO! do DNA repairs! because neutrophils (a type of immune cell) can attack s aureus with ROS (reactive oxygen species). these ROS loveee DNA its their favourite thing so they will go connect to DNA and cause single or double-strand breaks. and s aureus have a complex that fixes those on the spot.
yeah <3 thats it for your s aureus lesson.. go out into the world with this knowledge. do whatever you want with it
3 notes
·
View notes
Text
Also preserved in our archive (Daily updates!)
Published Sept 3, 2024
By Chuck Dinerstein, MD, MBA
New research reveals that fibrin, a key component of blood clots, may be the secret culprit behind the devastating neurological and inflammatory aftermath of the virus, including long COVID. From dense, stubborn clots to brain fog, the interaction of COVID’s spike protein with fibrin could be the missing link — and a potential target for life-saving therapies.
Coagulopathy, the formation of small blood clots that go on to wreck respiratory and neurologic havoc, has long been a clinical hallmark of COVID, and now it's oft-ignored Long COVID. A new study suggests that fibrin, a key component of blood clots, plays a role.
Fibrin provides structure to a blood clot and is derived from fibrinogen, a soluble blood protein when the coagulation cascade is activated. If you think of a blood clot as nature’s way of plugging a leak, fibrin deposition is frequently found where there is damage to the walls of blood vessels and the vessels making up the blood-brain barrier. Fibrin serves as a plug and a signal for a greater inflammatory and immune response.
Given the unique clinical presentation of clotting in COVID compared to other respiratory viruses, the researchers hypothesized that COVID directly binds to fibrinogen, promoting blood clot formation and altering clot structure and function. They found that the spike protein of the virus binds to fibrinogen and fibrin at specific binding sites, suggesting that the virus might contribute to abnormal clotting by interacting with fibrinogen.
They found that the spike protein altered the structure of clots, making them denser and more resistant to the body’s natural means of removing clots, a process called fibrinolysis. Additionally, the spike protein enhanced the inflammatory signals from fibrin, increasing oxidative forces (reactive oxygen species or ROS) released from macrophages, a first responder of the immune system.
In converting fibrinogen to fibrin, the spike's binding site (epitope) is exposed. Therapeutically, having identified binding regions, the research found that antibodies could disrupt and reduce these pro-inflammatory effects implicated in acute and long COVID. Among the inflammatory effects reduced by blocking the actions of fibrinogen was the deposition of collagen in the lungs, which creates a barrier to oxygen passage and helps to explain the refractory response to supplemental oxygen we have seen in patients.
Fibrin also suppresses natural killer (NK) cells, which are called "natural killers" because they can recognize and kill stressed cells without prior exposure to a particular pathogen, making them critical first responders. The suppression of NK cell activity results in enhancing viral persistence and lung inflammation.
In additional studies in mice, the researchers found that this fibrin-dependent inflammatory response occurs independently of the active virus, suggesting a potential mechanism for persistent symptoms in Long COVID. [1] Therapeutically, in their mouse model, the use of a monoclonal antibody targeting the fibrin epitope, in addition to reducing the lung’s inflammatory response, reduced neuroinflammation (associated with long COVID’s brain fog). There were reductions in fibrin deposition and microglial reactivity “leading to improved neuronal survival and reduced white-matter injury.” Microglia are the primary immune cells of the central nervous system.
To summarize:
Coagulopathy in COVID-19 is a primary driver of thrombo-inflammation and neuropathology rather than a consequence of systemic inflammation. Fibrin plays a causal immunomodulatory role in promoting hyperinflammation, neuropathological alterations, and increased viral load in COVID-19 by modulating NK cells, macrophages, and microglia. Elevated fibrinogen levels and BBB permeability in COVID-19 contribute to neuropathology, and targeting fibrin may offer a dual mechanism of action by inhibiting fibrin-spike interactions and exerting anti-inflammatory effects. A fibrin-targeting antibody effectively blocks many pathological effects of fibrin, providing neuroprotection and reducing thrombo-inflammation. Their findings have limitations, including how they measured changes in brain tissue, the use of mouse models, and the fact that our inflammatory response may have more than one pathway that results in COVID-19’s deleterious effects. For Long COVID, the fibrin-targeted antibody does not interfere with normal clotting, acting solely on fibrin's inflammatory responses, making it a candidate to protect against pulmonary and cognitive impairment; that will, of course, require clinical trials.
And there you have it—the silent saboteur behind the lingering specter of Long COVID. Fibrin is not just a bystander in the aftermath of COVID-19; it's a key player driving the chronic symptoms that continue to baffle patients and clinicians alike. The discovery that the virus’s spike protein meddles with fibrin, transforming it into a resilient, inflammatory force, opens a new frontier in the fight against the pandemic’s long tail. The research, though groundbreaking, is still in its early days, confined to animal models, and the complexities of human biology could introduce new challenges.
But if the science holds, targeting fibrin could offer a two-for-one punch against the clotting and inflammation that underpin much of the damage COVID-19 leaves in its wake. For the millions grappling with the enduring effects of Long COVID, this could be a glimmer of hope—a chance to reclaim their lives.
[1] The inquisitive with a conspiratorial bent might link these inflammatory responses in the absence of infection to deaths felt to be due to the COVID vaccines, which employ the spike as antigenic stimulus. The researchers note that most hematologic changes are triggered by the vaccine vector (an adenovirus) and that “COVID-19 RNA vaccines lead to small amounts of spike protein accumulating locally and within draining lymph nodes where the immune response is initiated, and the protein is eliminated.”
Source: Fibrin drives thrombo-inflammation and neuropathology in COVID-19 Nature DOI: 10.1038/s41586-024-07873-4 www.nature.com/articles/s41586-024-07873-4
#mask up#wear a mask#public health#pandemic#covid#covid 19#wear a respirator#coronavirus#still coviding#sars cov 2#fibrin#long covid#brain fog#chronic illness
77 notes
·
View notes