#Autosomal DNA
Photo

2 notes
·
View notes
Text

Patreon
#studyblr#notes#genetics#genetics notes#autosomal recessive#autosomal recessive disorders#genetic disorders#pathology#pathophysiology#disease prevalence#epidemiology#epidemiology notes#genetic diseases#prevalence of genetic disorders#biochemistry#dna
0 notes
Text
I've got a kind-of crack theory about Ruby's mother...
Back in The Church on Ruby Road, Ruby is invited onto Long Lost Family, a genealogy TV program hosted by Davina McCall, with the hope of finding some information about her bio family. Unfortunately, they come up with nothing.






[ID: 6 gifs showing Ruby and Davina McCall talking to each other on the phone from The Church on Ruby Road. Davina apologies to Ruby, who tries to hide her upset at the news.
DAVINA: "There is no trace of your mum or dad. I'm sorry. It happens sometimes."
RUBY: "No, that's fine... Thanks but, um, could you keep looking?"
DAVINA: "No, there's nothing more we can do. If your parents aren't on some kind of database, we can't find them."
RUBY: "Ok, um... isn't that unusual though? There's not a single trace anywhere? I mean... in the whole wide world, my mother's never left a blood sample or anythin'?"]
Now obviously, I know tracking down family is hard and, especially for orphans and adopted children, there's no gurantee that you'll be able to get the information you need. But I do find it odd there's seemingly "no trace" of Ruby's parents.
The section where I go on an odd tangent about genealogy
Speaking as someone who isn't a genealogist, but does enjoy researching family history in what little spare time they have... in my experience, close DNA matches aren't that hard to find. Especially if you're of white european descent, as Ruby is (presumably).
(It's generally harder for other ethnicities, as most research resources are white english/american focused. I know this is especially tricky for people like african-americans, where many of one's ancestors may have been enslaved. I've personally also found it tricky with Jewish communities as historically many of them used patronymic names prior to the 1800s, plus you have to account for immigration name changes, pogroms etc.)
For example, as someone who is white, with a mix of various british, mainland european, and ashkenazi ancestors, I actually have thousands of DNA matches, just from an autosomal test on Ancestry alone, let alone something like an mtDNA, xDNA or yDNA test:
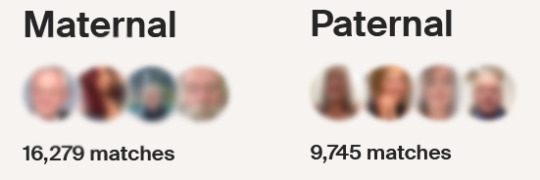
[ID: Edited screenshot showing maternal and paternal DNA matches on my AncestryDNA profile. There are 16279 maternal matches and 9745 paternal matches.]
Obviously, due to the way family trees work, most of these are distant matches, however it does include plenty of close ones too, which I've been able to trace to real records and identify relationships with. Personally, my matches even already include many 1st and 2nd cousins, albeit usually a one or two degrees removed, especially as the userbase tends to swing older on these websites. This includes a few people close enough for me to have already known them from family functions and shared annecdotes. Meanwhile, where I did have blank spots, from immigrations, estranged family members, early deaths etc, I've been able to fill in a lot of information.
So what does it mean that there's "no trace" of Ruby's family?
Deliberate or not?
The big question I've had since The Church on Ruby Road is: just how untraceable is Ruby's family?
On one hand, I feel like if this was real life and professional TV genealogists were helping you, you'd get a bit more information than a quick phone call saying they've got zilch. If they're sharing nothing... do they literally have nothing?
On the other hand, this also feels like a writing shortcut. We don't really need 3 hours of Davina McCall sat with Ruby at a computer breaking down every question and theory about possible family members. Ultimately, this was probably just a way to quickly get some major exposition out there, plus throw in a Christmas celebrity cameo for casual viewers. The fact they only talk about Ruby's "parents" being in a DNA database, and no-one else, doesn't give me a lot of faith in the care for accuracy RTD took with this plot point tbh.
Indeed Davina does say 'it happens sometimes', which could indicate it's not as extreme as having zero close relatives...
...but Ruby also asks if it's unusual for there to be no trace of anything, which Davina doesn't answer. If we're asking that question, it sounds like things really could have turned up that blank.
It may not be easy for orphans and adoptees to find family, but I assume it must be quite rare to have zero possible leads? Especially if you're a younger person, and thus may have a good number of people of the right generation to know/remember your family members still alive. Worst case scenario, I can imagine having some leads, only for someone to be uncontactable, or lack the information that would be useful. That being said, maybe I'm being too optimistic, as someone who had the priviledge of never having as much difficulty.
The weird sci-fi parallel (TW: incest (kinda), intersexism)
This is where we get to my theorising. Because in a science fiction context, and specifically a time-travel one, there is one quite famous short story that has a protagonist with zero family connections: '—All You Zombies—' by Robert A Heinlein.
(Fun fact: "All You Zombies" is also the name of a planned Class Ongoing story, once I get the time to resume that.)
You may also be familiar with the movie adaptation: 'Predestination'. It's also seemingly the inspiration for all sorts of similar stories, from 'The Man Who Folded Himself' to Red Dwarf and Futurama.
You might see where i'm going from that last one...
(Again disclaimer: if you seek it out, that this story may be quite triggering. It also was written in 1959. While it's actually somewhat respectable of a trans (kind-of, you'll see what I mean - I'll generally use the pronouns used in the text below) protagonist, it includes sexism, intersexism bordering on medical horror, and selfcest/incest.)
In 1963 (funnily enough), a lonely, orphaned 18 year old woman named Jane has a sexual encounter with a man in a park which ends up leaving her pregnant. When complications arise, the doctor discovers during a successful caesarian she's actually intersex, with a form of ovotesticular syndrome, with her immature, partially developed organs "a mess". He removes the now damaged womb, ovaries etc and, without consent, 'rearranged things so that [they] can develop properly as a man".
A few weeks later, the baby is stolen from the hospital by a man.
Despite all this tragedy, they do decide to complete their transition, restarting life as a man. He struggles to find work, but eventually finds himself making a living selling fake confession stories to magazines as "the Unmarried Mother".
Years later In a bar, he tells his story to a Bartender. After it all, the Bartender reveals he's actually a time agent and offers the chance to see his baby's father again. He drops him off in 1963 to find the man.
Meanwhile, in 1964, the Bartender steals a baby from a hospital, and drops her off at an orphanage in 1945.
The Bartender returns to the Unmarried Mother a month later in 1963, just in time to see him leaving a lonely young woman he met with in a park...
"Now you know who he is", the Bartender says, "—and after you think it over you’ll know who you are... and if you think hard enough, you’ll figure out who the baby is... and who I am.” He drops the Unmarried Mother off in 1983, where he can be recruited by the Temporal Bureau.
The Bartender, Jane, the Unmarried Mother, the kidnapper, the Father, and the Baby are revealed to all be one person, a family tree onto themself. The perfect time agent, causally disconnected from the rest of humanity and thus safe from Faction Paradox - if they are truly human at all (possibly explaining their biological bi-sexuality).
Thus, literally, having no relatives.
NO, OF COURSE I don't think this is what's up with Ruby!
But...
A lot of people have suggested that the woman who drops off Ruby could be herself. Obviously this doesn't necessarily mean Ruby is her own mother - let alone her own intersex father, child, and recruiter too!
But the story did come to my mind watching the Christmas special, and I do think the less squicky side of it, the 'perfect time agent' angle is worth considering. Could Ruby really be causally/genetically disconnected from the rest of humanity? Could she literally have no close relatives?
Assuming her DNA is not taken from any other person, but some semi-random mix of genes, she really may not match with anyone. At most, she would have some distant false matches, who share very small portions of DNA with her just by statistical fluke.
"BUT", I hear you say, "Didn't she get rewritten by the literal butterfly effect in episode one? She must be connected to humanity!"
Yes she did. But you know else happened?
She was still there.
Seriously think about it. Time travel fiction often doesn't think about the full consequences of time being altered even slightly, especially for a gag, but think about it literally. If all of human history was changed and a whole new species, possibly descended from Silurians, became dominant on the planet...
... why would the Doctor still happen to be travelling with someone with a name beginning with 'Rub-' who looks like Millie Gibson? Remember her name comes from Ruby Road... so does 'Ruby Road' exist on Rubathon's Earth? The Church presumably doesn't, unless there's a lizard Jesus...
At the very least we can point to the Web of Time being particularly reinforced around Ruby for some reason, even after all the damage it's taken between Flux and now, letting Ruby persist into the new timeline. This is explicitly confirmed in the last episode, with the Doctor calling it a fixed point.
At worst, it may imply whatever 'designed' Ruby just needs her to meet the Doctor, no matter what the dominant species on Earth is.
Mind you, both of these do open questions about what happened in the timeline where Ruby was eaten by the Goblin King. Maybe targetting her after her birth left her temporally vulnerable? Or maybe it was a necessary event, to bring the Doctor to Ruby Road...
Add this to some other things we've seen this season:
In Space Babies, we're introduced to the concept of 'baby farms', allowing people to be loomed born without a parent.
We also know, at least, that Ruby registers as human to the TARDIS (though given Sutekh's influence, who knows how trustworthy that scan was now!).
In The Devil's Chord, Ruby is not erased by Maestro destroying humanity. Granted we can put this down to the Doctor/TARDIS, and how time travel effects people's biodata, but I think it could be a misdirect.
(Interestingly there was a very similar plotpoint in "City of the Daleks", the Eleventh Doctor adventure game, which saw the New Dalek Paradigm invading Earth in...1963. Unlike Ruby, Amy eventually actually does start to fade, needing a 'chronon blocker' to stabilise her. Hey remember how we just heard the word 'chronon' used a bunch in the show.)
In Boom, the Ambulance is entirely unable to find a next of kin for Ruby, despite seemingly having her in its records. This is a little hard to dissect, as you could take a lot of different interpretations away from it. At the very least, it suggests Ruby doesn't have any living descendents in the 51st century. Carla probably doesn't either (which makes sense with her not having any bio-kids, and Ruby seemingly being the only child she fully adopted rather than fostered?) But for its extensive records, it's notable it still couldn't find anyone after that, even presumably with access to Ruby's DNA like the genealogists had.
Everything in 73 Yards.
Between the snow falling in each episode, plus context in The Legend of Ruby Sunday, we know that Christmas Eve on Ruby Road, while fixed, is also uniquely vulnerable and 'raw'. With the woman's changing reactions to the Doctor, it's also flexible enough to change, somewhat.
Similarly, the possible connection between the woman who dropped Ruby off and the woman in 73 Yards, between her face not being visible and the CCTV camera being around 73 yards / 66.6 metres away. And if that woman really was Ruby, then maybe the parallels to All You Zombies may not be as insane as they sound.
#Doctor Who#DW Spoilers#Doctor Who Spoilers#Ruby Sunday#The Legend of Ruby Sunday#All You Zombies#DW Theory#DW Meta#long post
93 notes
·
View notes
Text
ok i saw the term "jewish dna" again today and im sorry but this is not how dna works
it's not like each ethnicity has a "type" of dna and if you have that you're that thing
how dna testing companies that tell you about your ancestry work is this:
every so often there are changes to dna that occur, like mutations and deletions. those then get passed down to your kids. let's say you had a mutation on a specific gene, and then someone does dna testing on your kids and your sister's kids. they'll see that your kids had the mutation and your sisters' didn't, right?
so basically dna testing for ancestry does that on a huge scale. they can tell that you're part of a group that branched off from some other group at some point in history. that way they can tell which groups are genetically related to each other.
so they can use this to tell that someone has jewish ancestry because they're seeing which mutations, deletions, etc you have and that they fit into various branches of jews.
but that doesn't mean that the dna is itself "jewish" in any meaningful way. also the branches, called haplogroups, often encompass larger portions of the population. for example i am in maternal haplogroup K, which is super common among ashkenazi jews. but there are also a lot of non-jews (in the middle east and southern europe) who are part of haplogroup K, as well as many ashkenazi jews who aren't.
if i hadn't been raised knowing i'm jewish and i wanted to find out, we'd still be able to find that out using dna testing. we'd be able to use the maternal dna data, plus my autosomal dna (the ones on the non-sex chromosomes) to look at other branching history and we'd piece it together.
but im not jewish BECAUSE of the dna. rather it's a tool we can use to find information.
44 notes
·
View notes
Text
When it comes to choosing my favourite detail in Milgram, then Yuno's hair clips are definitely it!!


The hair clips together form an X and a heart!
The fact that there are two hair clips and one of them has an X shape, it really looks like a pair of chromosome!
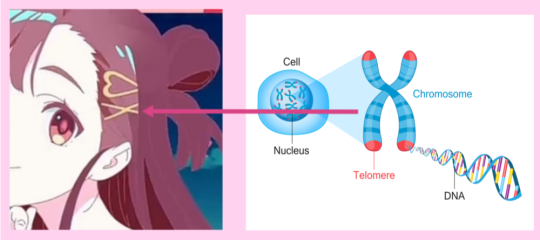
"Chromosomes are bundles of tightly coiled DNA located within the nucleus of almost every cell in our body. Humans have 23 pairs of chromosomes."
"Humans have 23 pairs of chromosomes (46 in total): one set comes from your mother and one set comes from your father."
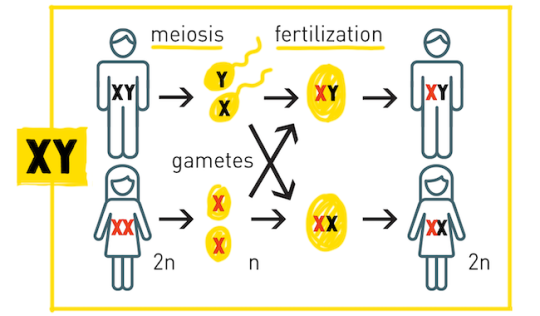
"Of these 23 pairs, one pair are sex chromosomes so differ depending on whether you are male or female (XX for female or XY for male).
The other 22 pairs are autosomes (non-sex chromosomes) and look the same for both males and females."
The heart although it could be just a simple, cute accesory, might also symbolise that Yuno wasn't aware of the baby's sex, since the first one is always X and the second one changes: Y - male, X - female. The heart could be like an unknown variable!
It might also imply that Yuno was just 1 or 2 weeks pregnant at the time, since the zygote (which has the chromosomes which determine the baby's sex) is not formed until the third week.

And... That's all! Just thought it was interesting to mention!
34 notes
·
View notes
Text
I’m so excited for all the classes I’m starting this fall <33
List if anyone’s interested:
- intro to archaeology (required for my major, my friends who took it last year said it was their fave of all the required Anth courses— I’m a bioanth girlie but I’m sure I’ll have fun)
- intro to genetic anthropology (we get to take an autosomal dna test and learn how to analyze the data!!)
- Ancient Greek 101
- Russian 1
It’s only four classes but it is 14 credits total (for reference 12 is full time), which is kind of my sweet spot when it comes to getting things done at a good pace without being overwhelmed.
9 notes
·
View notes
Note
BIO QUESTIONNNNS
any tips on interpreting pedigrees???
How does fermentation work 😭
whats your favourite organelle??
GL on the exam!!!!
THANKKK YOUUUU FORR THE ASKKK
Pedigrees
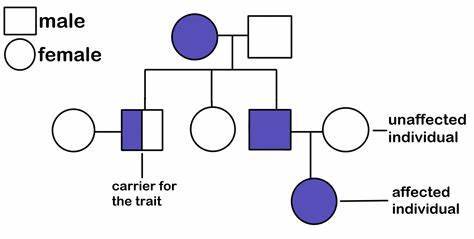
these guys were sooooo puzzling when I first looked at them. But once you get a feel for them, they aren't too bad. (Then they become a fun puzzle and you feel so smart and smug /pos)
Sometimes they'll include the half-shaded shape, sometimes they won't. On my practice exams they don't so I won't include talking about the half-shaded shapes (which are basically like telling you which ones are carriers, when nothing is half shaded then you have to figure this out yourself.)
Okay so first off, I typically look for sex-linked/autosomal traits. If the affected individuals are usually one sex (male typically) then the trait is sex-linked. If there's siblings and only the males (typically) are affected, then again - sex linked. (I wishhh we could learn biology with intersex people included it wouldd bee soo nicceee)
ALSO ALSO, if the mother is affected, then all of their children are affected - no matter what the trait of the male parent is - then I'd think the trait is passed on through mitochondrial DNA. (mitochondrial DNA is sooo coooool I need to reread those pub med articles. But overall, mitochondrial DNA comes from the female parent only because the male parent contributed only Nuclear DNA, the female parent contributed the cytosol and every organelle in it.)
From there, if neither of the above is true, then it's typically autosomal and either a recessive or dominant trait.
If it's autosomal recessive, then typically it will skip generations/ only some offspring will have it. It's just less likely that the recessive trait will appear, bonus points if there's four siblings and one of them is affected. (Not reallllyyy how it works in the real world since genotypes for each child is independent from the other children... but IDK pedigrees on tests and stuff tend to do this.) Also if none of the parents have it but some children are affected, then one or both of the parents are most likely carriers. (and if you can see the parents of the parents, you can typically find out which one).
Dominant ones just... tend to appear more. They don't usually skip generations, and if neither parent is affected, then the children won't be affected. (You can't be a carrier for a dominant allele under most circumstances... not sure if there's exceptions)
Pedigrees take a bit of getting used to, but they are FUN once you get a feel for them. (Tbh our ap bio class is kind of cursed, we went through a few teachers that all quit. Now I'm the honorary teacher of our class and honestly... I just got a feel for them. Practice I suppose? If you remember how different traits are inherited, then you should be good) Good luck with them!
Fermentation
OKay so not going to lie here.... I usually skipped over fermentation because I thought it would be boring. Once I saw this ask, I looked at it and it's... surprisingly not that bad? (At least to me, I'm insane.)
Okie so basically what happens is
~oxygen~ the most important molecule for us, breathe they say, breath is the language of the soul, our almighty oxygen...
... mops up the waste products at the end of the electron transport chain in oxidative phosphorylation (hydrogens) and turns into water.
But, despite how unromantic that sounds... it's super important. Because without our oxygen janitors, we can't do the electron transport chain. The waste products would build up and we'd probably burst into flames or smth (honestly that is what would happen if our cells tried to metabolize glucose all at once)
So cells that do fermentation decide to forgo the whole electron transport chain, best not deal with it.
So all it has is glycolysis. (I call it the carbon do-see-do, whatever that dance thingy is called. I don't know why.) I'll include a picture here of it
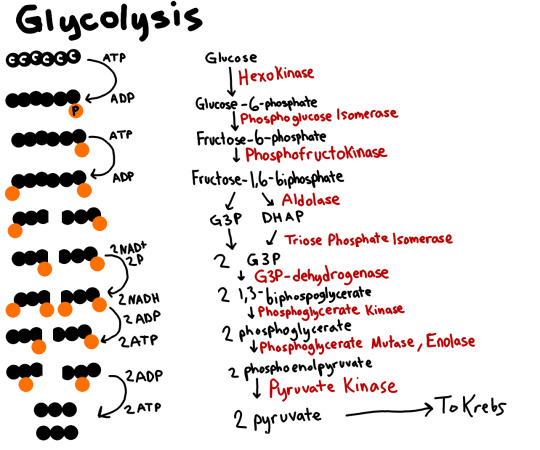
(knowing the names of the intermediate products is probably the hardest part.. I just... don't.)
Basically, add your phosphates to each side, split it in half, add another phosphate (not from ATP this time), Oxidize the heck out of the carbon molecules, then give those electrons to the NAD+, which turn into NADHs (One per half/three carbon molecule.), then you break off your 'caps' of Phosphates and add it to ADPs. ONce you add that extra phosphate, you make the ADPs into ATPs. Four of them total, but because you invested two ATPs, you got two 'new'/net ATPS.
I'll explain a few more concepts because I think I can do better than the textbooks if I do say so myself. (Especially the ones I got for school, mc graw hill ones.... like... ehhhhh it's so weirdly written. The book I read when I was little is called 'the way we work' gorgeous pictures and lots of metaphors, definitely one of the things that got me into biology, and helped me understand it.)
Adenosine triphosphate is basically a kitkat. Lots of energy stored within the last three phosphates on the side. (which slightly repel each other, so there's a lot of pent-up energy stored, which is why it takes all of cellular energy to make these, you have to go against the natural tendencies. The plus is that, like a kit kat, they are easy to break and release a lot of energy)
If you look at biochemistry, signal transduction pathway, transportation across the cell membrane, you use these phosphates (very negatively charged) to power EVERYTHING. Think of using an explosion to power your solar panels so you can make something. Like that. So when you do glycolysis, you're just adding those phosphates onto the ADPs to 'remake the Kitkat'/ put two negatively charged magnets together.
Basically, you're energy harvesting. The electrons hold energy, you take that energy from the glucose to give it to the NADH, your electron carrier.
HOwever, you can't do the electron transport chain. So now what do you do with the energy...
Discard it. You got two ATPs, and without oxygen, that's the best you're going to get. (Compared to around 38 ATPs with oxygen. Helpful little janitor.)
Essentially, you have pyruvate (the 3-carbon molecule you made from splitting glucose in half in glycolysis) and two NADHs that you need to turn back into NAD+ so you can do glycolysis again.
(IDk why I'm using second person now. By 'you' I mean the cell. ANd by the cell I mean I'm personifying, no one really 'wants' anything here. It just... happens.)
To do this, fermenting cells have this little trick, they basically give the electrons back to the pyruvates. Return them from whence they came. (apparently there's a good return policy)
If it's a muscle cell or yogurt or the likes, the NADHs give the energy/elections back to the pyruvate, which turns it into lactic acid.
If it's yeast, the NADHs give the energy/electrons back to one of the intermediate products of glycolysis. (I forget the name). This produces ethanol and CO2
So now you got your NAD+'s back. And you have two ATPs (net, four total). Not the... not the best but it's what you got.
OKIE DOKIE THAT'S IT. I RECCOMEND THE AEOMBA SISTERS IF YOU STILL NEED HELP /pos.
Favorite organelle
akldfjalskdfj tha's like askingg meee to chosee a favoritee ficitonall characterrrr.
..a jdflaskdfjaskdlfjaslkdfj
I can't pick favorites for anyythinggg.
LIke the ER does protein production, but the nucleus has the DNA and such, and of course we have the mitochondria (the powerhouse of the cell they say), and the Golgi body post office and the lysosome slaughterhouses and the centrioles with their cytoskeleton and their miotic spindle.... you can't do this to me /pos.
kladsfjasdf ENDOPLASMIC RETICULUM BECAUSE THE CELL MAKES PROTEINS THERE. kafjkaadlfkjaks AAAAA-
THank you for wishing me luck! I... I hope I don't ramble on the free response questions...or use metaphors... ha.
(also also, haven't really storyfied cellular respiration because...twas hard to storyfy. THey don't include the enzymes involved because it's too complicated. I tried my best!)
#cellular biology#biology#science#science side of tumblr#science side please explain#science side help me#science side of the internet#science side explain#pedigree#fermentation#lobotomy for my brainrot#noorie infodumps. be very afraid#noww do you all understand that tag? /pos
10 notes
·
View notes
Text
This evening, I will adopt an academic perspective to deconstruct the misleading notion that human differences are primarily attributed to skin color or variations in pigmentation. It is fundamentally misguided to consider blood as a determinant of racial purity, especially when scientific understanding reveals that humans can be categorized into a mere four blood types, which can expand to eight when considering the Rh factor.
1. The 20 amino acids discussed are all L-isomer, α-amino acids. Chromosomes, which are threadlike structures composed of proteins and a single DNA molecule, function to transmit genomic information between cells. In both plants and animals, including humans, chromosomes are located within the cell nucleus. Humans possess 22 pairs of autosomes and one pair of sex chromosomes (XX or XY), totaling 46 chromosomes.
2. Each of the four nucleotides—adenine (A), thymine (T), guanine (G), and cytosine (C)—is formed by attaching a phosphate group and a nucleobase to a sugar molecule. The sugar present in all four nucleotides is deoxyribose, characterized by its cyclic structure, which consists of one oxygen atom and four carbon atoms arranged in a ring. Additionally, a fifth carbon atom is linked to the fourth carbon in the ring, and a hydroxyl group (-OH) is bonded to the third carbon.
3. This underscores the undeniable reality that all humans share a commonality, challenging the notion that individuals with red or yellow skin differ fundamentally, or that the concept of whiteness supersedes all human classifications; in truth, no human can reproduce the full spectrum of skin color variations except for those identified as Black.
2 notes
·
View notes
Text

🇮🇱 … Israel is nothing but a European colony implanted in the Middle East! —.No one wanted these ‘settlers’ and the a British decided (after considering several other ‘lands’) to send them to Palestine!!
Notice the quote by Arthur Khosla!!
By: LaillaB, founder of ‘Reclaim the Narrative’, from LinkedIn …
“Israel should be viewed as a European colony implanted in the MiddleEast rather than having genuine native semitic roots to the land argue various scholars and academics.
A colony with a story rooted in colonialism, a project that has slightly modernised its visage but continues to subject Palestinians to military occupation and land dispossession.
🔬 Autosomal DNA admixture of Palestinian Christians, Muslims and Ashkenazi Jews; National Library of Medicine 2010:
🔻Palestinians derive most of their ancestry from the Bronze Age
🔻Canaanites, 83% among Christians and 70% Among the Muslims.
🔻Ashkenazis derive most of their ancestry from a Southern European source and not the Canaanites.
🔻Closest modern groups to Palestinians; native Levantine groups, Lebanese and Jordanians.
🔻Closest modern groups to Ashkenazis are Europeans.
Genealogically speaking, & politics aside, Ashkenazis would be considered outliers and not Native to the Levant Area.
All the Levantine Arabs, including Palestinians, have a common genetic inheritance in continuity with the Canaanites.
Palestinians are for the most part descendants of groups who have been in the Middle East for 4 thousand years, at least.
Despite the significant historical dna ties (roots), the coloniser continues colonising Semites, resulting in the massacre of over 40,000, which no longer warrants the term "plausible” genocide.
Israel's imperial ambitions in their current ethnic cleansing campaign of Gaza, driven by the desire to exploit the region's natural resources and fulfill their vision of constructing the Ben Gurion Canal.
This project aims to rival Egypt's Suez Canal and establish (lucrative) strategic trade dominance, underscoring Israel's colonial agenda driven by exploitation;
According to Israeli historian Ilan Pappe in his book “On Palestine”, 2015 co-written, American scholar Noam Chomsky:
“The tale of Palestine from the beginning until today is a simple story of colonialism and dispossession, yet the world treats it as a multifaceted and complex story, hard to understand and even harder to solve.”
Malcom X argued in his article, “Zionist Logic” …
“Did the Zionists have the legal or moral right to invade Arab Palestine, uproot its Arab citizens from their homes and seize all Arab property for themselves just based on the “religious” claim that their forefathers lived there thousands of years ago? Only a thousand years ago the Moors lived in Spain. Would this give the Moors of today the legal and moral right to invade the Iberian Peninsula, drive out its Spanish citizens, and then set up a new Moroccan nation . . ., as the European Zionists have done to our brothers and sisters in Palestine?” 1964.
“Does Israel’s existence as a Coloniser, an Occupier, a Settler Colony have any claim or any right?
Not historically, not biblically, not logically and not legally. Religion is no basis to claim a right to inherit another land.
Call it what it is!! … 🇮🇱
#reclaimthenarrative —🕊🍉 — #FreePalestineFromTheRiverToTheSea … @hrexach …
#dr rex equality news information education#graphic source#graphic#graphics#hortyrex ©#horty#quote#it is what it is#palestine#israel#israhell#fake#european countries#colony#colonialism#oppression#the dispossessed#history#british#uk#racism#injustice#the imposition of unnecessary obstacles
2 notes
·
View notes
Text
Sunday Six
“I do hope, if it is the case, I’m at least in the running for godmother.“
“I already asked Ivy. However,” Oswald hedges, “any child in Gotham can use more than one adult to count on.“
—
Barbara, it turns out, isn’t in the running for estranged mother either: the mitochondrial DNA isn’t a match for any of the living Arkham patients and permission to run tests from deceased patients like Jane Cartwright are months away. They’re luckier with the autosomal DNA — even with half a dozen cross matches. Baby Etheline has relatives on Oswald’s side, on Jonathan Crane’s side but the closest paternal match is Edward Nygma. Who, in a move that shocks even her, disappears.
4 notes
·
View notes
Text
Future Child Diagnosed with Huntington's Disease
Hi everyone, I am 4.5 months pregnant and just got a pre-natal genetic test done which revealed that the child I am carrying will be diagnosed with Huntington's disease. I don't completely understand everything just yet, so if anyone with similar experiences has any advice, I'd really appreciate it! Here is what my doctor said and what my own research has amounted to so far:
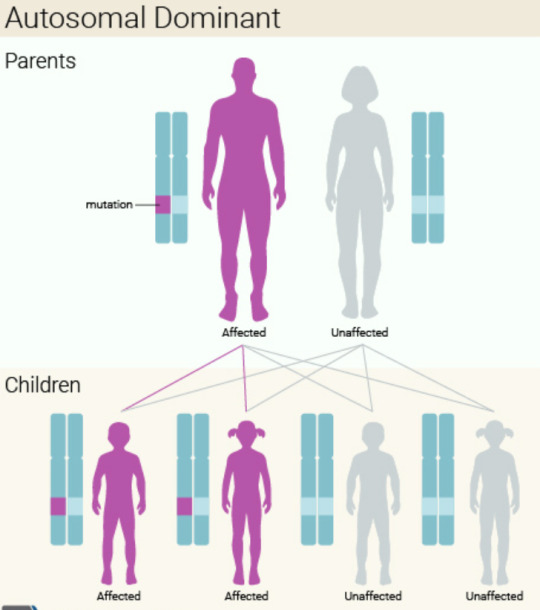
How my child inherited it unknowingly: Huntington’s disease is an autosomal dominant disorder, which means that a person only needs one copy of the gene to develop the disorder. It is caused by mutations in the HTT gene. The HTT gene provides the instructions for making a protein called huntingtin, too much of the protein is toxic to nerve cells. The HTT gene has a DNA segment known as the CAG trinucleotide repeat which is usually repeated 10 to 35 times. In people with Huntington’s disease, the segment repeats 36 to more than 120 times, however, those whose segment repeats 36 to 39 times may not develop symptoms. My husband has the gene and his CAG segment repeats 37 times, but he thankfully won’t develop symptoms as he’s already reached the general age of development, his 30s or 40s, however, he has passed down the gene to our future child.
Symptoms:
Causes the degeneration of nerve cells in the brain.
Affects functional abilities and usually results in movement, cognitive, and psychiatric disorders.
Movement disorders: chorea (involuntary jerking or writhing movements), dystonia (muscle problems such as rigidity or muscle contracture), slow or unusual eye movements, impaired walking, posture, and balance, and difficulty with speech or swallowing.
Cognitive disorders: difficulty prioritizing or focusing on tasks, tendency to get stuck on a thought, behavior, or action, lack of impulse control, lack of self-awareness, slowness in processing, and learning difficulties.
Psychiatric disorders: feelings of irritability, sadness, or apathy, social withdrawal, insomnia, fatigue, suicidal thoughts. OCD, mania, or bipolar disorder.
Causes of death: pneumonia or other infections, injuries related to falls, complications related to the inability to swallow.
Treatment: The disease is unfortunately not curable, but medications are available to help manage the symptoms. Medications include drugs for movement control such as tetrabenazine and deutetrabenazine, but they may cause drowsiness, restlessness, and the risk of worsening or causing depression or other psychiatric conditions. Antipsychotic drugs such as haloperidol and fluphenazine may be beneficial in treating chorea but may worsen dystonia, restlessness, and drowsiness. Quetiapine and olanzapine may help suppress violent outbursts and other mood disorders, however, may cause movement disorders themselves. Antidepressants such as citalopram, escitalopram, fluoxetine, and sertraline may also treat OCD, however, may cause nausea, diarrhea, drowsiness, and low blood pressure. There is also psychotherapy, speech therapy, and physical therapy.
There also seems to be some current research being done by scientists. They are trying to understand the toxicity of the mutant huntingtin protein and how to develop a potential drug to counteract it. Scientists are using cutting-edge methods (hope it works!!) such as optogenetics (neurons activated or silenced in animal brains using light beams) to study circuit defects in Huntington’s disease and are using stem cells to study disease mechanisms and to test potential therapeutic drugs. My child will have many years until the symptoms start so I hope that the research will become tangible enough to relieve them of their future symptoms.
This is all really just scientific research so if any other parents in the community have any advice, please post it!!
5 notes
·
View notes
Text
HEREDITARY FORM OF EPILEPSY ASSOCIATED WITH PYRIDOXAMINE 5'-PHOSPHATE OXIDASE DEFICIENCY IN A CHILD by Plotnikova I.A in Journal of Clinical Case Reports Medical Images and Health Sciences
SUMMARY
The article presents a clinical case of focal epilepsy with a status course of seizures associated with a genetic mutation in exon 1 of the PNPO gene, which led to pyridoxamine-5'-phosphate oxidase deficiency. The diagnosis was made late due to the misinterpretation of symptoms, which complicated the course of the disease. Despite the fact that the first symptoms in the form of seizures appeared at the age of 1 month, only at the age of 5 the diagnosis was verified by doing targeted DNA sequencing. At the moment, the patient is receiving substitution therapy in the form of pyridoxal phosphate 300 mg/day, which enabled unstable clinical remission. Right now, it is impossible to achieve complete control over the convulsive syndrome without a strict diet: dairy-free, meat-free, egg-free and low-protein fat-free food. Currently, further search for treatment methods continues to improve the patient's quality of life and ensure stable remission. A detailed analysis was given for further genetic verification based on the amino acid profile of the patient, and the rehabilitation potential was determined based on topical neuropsychological diagnostics performed on a non-verbal child.
Key words: focal epilepsy; Pyridoxal 5′-phosphate; vitamin B6; PNPO; vitamin B6-dependent epilepsy, neuropsychological diagnostics.
INTRODUCTION
Vitamin B6-dependent epilepsies are aheterogeneous group of autosomal recessive diseases that are caused by mutations of five different genes involved in vitamin B6 metabolism [1]. Vitamin B6 is present in many forms in the human diet, but only pyridoxal-5 -phosphate (PLP) plays a vital role in the metabolism of a number of neurotransmitters, especially the inhibitory mediator gamma-aminobutyric acid. Code errors leading to a lack of pyridoxal-5'-phosphate manifest as B6-dependent epilepsy, including pyridoxamine-5-phosphate oxidase (PNPO) deficiency, which affects the synthesis and recycling of pyridoxal-5'-phosphate [2,3]. Neonatal manifestation in the form of acute encephalopathy with biphasic epileptic seizures (or status epilepticus) is the main symptom of the disease. The first phase (early attacks) is accompanied by fever and a temporary recovery of consciousness and the development; the second phase is a global cognitive dysfunction (late attacks).
Resistance to traditional antiepileptic therapy requires patient's lifelong treatment by pharmacological doses of vitamin B6 in the form of pyridoxine (PN) or a biologically active form of pyridoxal-5’-phosphate [1,4].
Case reports of PLP deficiency, verified not only clinically, but also by exome sequencing, are quite rare as well as the methods for studying molecular markers of alpha-aminoadipic semialdehyde and pipecolic acid in body fluids [5–7]. The complexity of diagnosis is caused by multiple disorders in newborns, especially in case of a slow and incomplete response to pyridoxine [8].
Recent studies have shown that the main enzyme defect in pyridoxine-dependent epilepsy is caused by alpha-aminoadipic acid semialdehyde dehydrogenase in the pathway of cerebral lysine degradation. The accumulating compound, alpha-aminoadipine semialdehyde (alpha-AASA), is in equilibrium with delta-1-piperidine-6-carboxylate (P6C). P6C inactivates pyridoxal-5’-phosphate, causing severe cerebral insufficiency. Although treatment of pyridoxal 5'-phosphate deficiency can successfully control seizures, most patients develop some degree of disability, regardless of early diagnosis and treatment. Very few patients with normal intelligence have been reported [7].
Objective: to analyze the course of epilepsy with pyridoxamin-5’-phosphate oxidase deficiency in an 8-year-old patient with diagnosis verification by clinical exome sequencing.
MATERIALS AND METHODS OF RESEARCH.
The analysis of primary medical documentation from 2013 to 2021 of a patient born in 2013 was performed. We reviewed the materials on the topic using PubMed search engines for the period 2014-2021, correlation of literature data with a specific clinical case.
RESEARCH RESULTS AND THEIR DISCUSSION.
A clinical case
Girl, 8 years old, was born from IV pregnancy of a woman with a burdened obstetric history. At the age of 1 month, tonic-clonic convulsions were first noted during sleep: gaze adversion to the left, lasting 30 seconds - 1 minute; afterwards there was up to 4 seizures per day, daily. At the age of 1 year, she was hospitalized 4 times on an emergency basis for convulsive seizures. The child was observed by a neurologist-epileptologist with a diagnosis of perinatal damage to the central nervous system, recovery period. Valproic acid was prescribed at a dosage of 50 mg/kg per day, oxcarbazepine 300 mg/day, without an effect of therapy. At the age of 2 years, she was hospitalized three times in the intensive care unit due to the status course of an epileptic seizure with a rise in temperature to febrile numbers. Neurological diagnosis at that time was: symptomatic epilepsy with complex partial seizures, status course of generalized convulsive seizures. On electroencephalography (EEG): moderate diffuse changes in the bioelectric activity (BEA) of the brain in a disorganized type. The patient's condition worsened. At the age of 3 years, she was observed in the State Autonomous Healthcare Institution of the Sverdlovsk Region "Children's City Clinical Hospital No. 9, Yekaterinburg" with the same diagnosis; the dose of oxcarbazepine was increased to 500 mg/day, valproic acid to 300 mg/day with no significant clinical effect. At the age of 4 years, she was hospitalized three times in the intensive care unit about epileptic seizures, without the effect of anticonvulsant therapy. Concomitant diseases at age 4 were: severe osteoporosis of the visible parts of the skeleton; pathological compression fracture of the body Th11; hepatomegaly; moderate expansion of the common hepatic, common bile ducts; enlargement of the gallbladder; a pronounced increase in the size of the kidneys, pancreas; diffuse changes in the parenchyma of the kidneys, a single cyst of the right kidney; unspecified form of caries; chronic gingivitis. Computed tomography of the abdominal aorta and its branches showed no evidence of hepatic artery stenosis. Autonomic dysfunction of the sinus node was noted: sinus arrhythmia with episodes of bradycardia. There were also small anomalies in the development of the heart: a functioning foramen ovale, additional chords of the cavity of the left ventricle.
DNA sequencing was carried out in 2017. Genetic mutations that were identified are described in patients with epilepsy associated with pyridoxamine 5'-phosphate oxidase deficiency and, based on the totality of information, regarded as pathogenic - a mutation in exon 1 of the PNPO gene (chr17: 46019139A> T, rs370243877), leading to amino acid replacement at position 33 of the protein (p.Asp33Val, NM-018129.3, mutation frequency in the ExAC control sample 0.0235%); as probably pathogenic - a previously undescribed heterozygous mutation in intron 3 of the PNPO gene (chr17:46022086G>A, rs766037058), leading to disruption of the splicing site and synthesis of the full-length protein (c.363+5G>A, NM_018129.3, OMIM: 610090, the value of the algorithm for predicting its influence on the function of AdaBoost splicing sites is 1.000).
A heterozygous mutation was also found in exon 4 of the EARS2 gene (chr16:23546678A>T), leading to a premature translation termination site at codon 163 (p.Tyr163Ter, NM_001083614.1). Such mutations have been described in patients with combined oxidative phosphorylation deficiency type 12 (OMIM: 614924). In this case (when no second mutation in the gene is detected), the result is regarded as an option with uncertain clinical significance, however, the mutation may be related to the phenotype. The parents did not undergo a genetic examination.
Prescribed treatment was: pyridoxine hydrochloride intramuscularly, then - pyridoxal phosphate at the rate of 10-50 mg / kg /day. On the 7th day after the start of treatment, the patient's consciousness was assessed as clear, she was able to sit up independently and stand with support. Her seizures stopped, appetite improved, during rehabilitation positive dynamics in neuropsychic development was noted with an expansion of the range of motor activity, the appearance of gaming activity, emotions and attempts to pronounce individual sounds.
At the age of 5 years 1 month there was a new epileptic seizure. The dose of pyridoxal phosphate was increased to 600 mg/day, convulsive attacks stopped. Concomitant diseases at age 5 were perianal dermatitis, vulvitis, continuously recurrent leukocyturia. Subsequent courses of medical rehabilitation was prescribed with positive dynamics.
In 2019, hyperkinesis (blinking), tremor, restlessness reappeared; in the summer were tonic-clonic seizures with vocalization, lasting 15-20 minutes and the status course of an attack, operculations, loss of appetite. By the end of the year, there was constant nausea and a gag reflex at the sight of food, vomiting with yellow mucus and a sour smell once every 5-7 days, accompanied by febrile fever, the smell of "rotten cheese" from the scalp and excrements during attacks. Motor clonic seizures appeared with a frequency of once every 1-2 months, symmetrical chill-like tremor - up to 3-5 times a day. Periodic episodes of psychomotor agitation, stereotyped movements were also noted.
Neurological status. There are bradypsychia, delayed psycho-motor development, coordination disorder. Patient does not pronounce words, speech is active only during the game-vocalisms, self-service skills are not formed. Autism spectrum disorders with general speech underdevelopment of level 1, psychomotor alalia were noted. Cerebral, meningeal symptoms are negative. The gait is uncertain. Cerebellar tests are negative. Cranial nerves: palpebral fissures D=S, pupils D=S, pupil reaction to light: direct D=S, consensual D=S. The volume of movement of the eyeballs is complete D=S, there is no nystagmus. The face is symmetrical D=S. There is no language deviation. Swallowing, phonation are not disturbed. Muscle tone: arms - reduced D=S, legs - normal D=S. Tendon reflexes: from the arms and legs increased D=S. There are no pathological foot signs, pelvic functions are preserved. Patient shows signs of slightly asymmetrical (with an accent on the left) motor awkwardness, reduced nutrition (Body weight 21,5 kg).
Results of instrumental and laboratory studies. The following disorders were detected on the EEG prior to the start of etiological therapy: Epileptiform activity in the form of "peak-wave" complexes in the frontal and central-temporal leads, more on the right; slowing down of activity in the temporal zone.
In the biochemical analysis of blood the level of amino acids (µmol/l) is low: alanine 119.30; glutamic acid 72.00; glycine 86.50; ornithine 22.10; proline 87.00. Activity of alanine aminotransferase is 24.9 U/l (reference values 0-29 U/l), aspartate aminotransferase - 26.4 U/l (reference values 0-48 U/l).
Control visit. After the diagnosis was verified by exome sequencing, the patient was prescribed etiotropic therapy: pyridoxal phosphate 300 mg/day. The pre-elevated (1070 nmol/l) plasma concentration of vitamin B6 (pyridoxal-5-phosphate) normalized. EEG data - video monitoring showed moderately severe violations of BEA of the brain; the main rhythm is formed by age; registered regional slowing of the rhythm in the right central-parietal region. Epileptiform activity, clinical paroxysms, EEG patterns of epileptic seizures were not registered.
Final diagnosis: Genetic focal epilepsy due to a mutation in the PNPO gene (chr17: 46019139A> T, rs370243877). The type of attack is focal with impaired consciousness. PNPO developmental and epileptic encephalopathy. Cognitive impairment. Alalia. Motor awkwardness.
Psychological status. Diagnostics of cognitive activity showed that the girl is accessible to contact; she does not speak and comprehension of the speech is shown only in the form of understanding simple commands and simple instructions for the task. The child's object-sensory activity is carried out 100% through visual perception and shape perception, the perception of size is developed by 50%, spatial perception - 12%, color perception is completely absent. The insufficiency of these afferentations is a consequence of the decrease in the “zone of actual development”, which may be attributed to pedagogical neglect. In the motor sphere, gross motor skills are fully formed, fine motor skills are developed by 54%, objective activity is formed by 9%, taking into account the skills of game and constructive praxis, speech function is developed by 25%, self-service skills - by 60%, socialization – by 40%. Psychological diagnostics of the state of higher mental functions was carried out by depicting the structural and functional features of the brain, as a result of which topical insufficiency of brain areas was revealed. Figure 1 shows the level of formation of brain zones.
Figure 1: The degree of formation of brain departments that implement sensory and motor skills.
Despite the pronounced cognitive deficit in the child, the implementation of the program of psychological rehabilitation may expand the "zone of actual development" in the structure of the sensory, subject and pedagogical profile (since there are preserved components of cognitive activity)
DISCUSSION
Patient’s clinical diagnosis was established only at the age of 5 years, based on clinical manifestations and exome sequencing. The primal reduction of the dose of pyridoxal-5'-phosphate provoked a relapse of status epilepticus and a regression of acquired cognitive skills. A subsequent increase of treatment in combination with dietary therapy provided an unstable clinical remission without further improvement in the patient's condition. Such a response to the therapy has also been demonstrated in other studies [6,7].
Although in patients with a typical course of the disease, there is a several-fold increase in the level of glycine and glutamic acid in the blood plasma [1,5–7,9], in our case there is a decrease in glycine to 86,50 µmol/l (norm: 100-400 µmol /l) and other amino acids. Hypoglycinemia is an extremely rare condition, it occurs only in severe hereditary aminoacidopathy, but in our patient, tandem mass spectrometry was performed twice (including against the background of an attack) in 2016 and did not show any data of hereditary aminoacidopathy, organic aciduria, defects β-oxidation of fatty acids. The girl has a positive reaction to the oral intake of amino acid complexes and glycine separately, therefore, additional genetic analysis can be performed for 3-phosphoglycerate dehydrogenase deficiency, the clinical manifestations of which may be encephalopathy and seizures unresponsive to anticonvulsants [10]. Symptoms of this disease can be stopped by joint intake of serine and glycine so this diet may be developed for our patient. The study of vitamin B6 metabolites in de novo serine biosynthesis by Ramos et al (2017) had one group of rats which received a pyridoxine-deficient diet, while the diet of the control group of rats contained a normal amount of pyridoxine. This study has demonstrated a decrease in serine biosynthesis in Neuro-2a cells in vitamin B6 deficient rats. The pyridoxal-5'-phosphate-dependent enzyme phosphoserine aminotransferase (PSAT, EC 2.6.1.52) cannot function fully in conditions of vitamin B6 deficiency, and likely reduces the synthesis of phosphoserine and serine in animals on a pyridoxine-deficient diet. The production of glycine depends on the availability of serine and on the pyridoxal-5'-phosphate-dependent enzyme SHMT, which catalyzes part of the transformation of glycine, and the simultaneous deficiency of serine and pyridoxal-5'-phosphate can reduce its activity and lead to a decrease in the content of glycine in blood plasma [9].
Some authors reported EEG changes in patients with pyridoxine-dependent epilepsy [11]. In our patient, no clear epileptiform activity was registered either before or after the start of treatment with pyridoxal-5'-phosphate; this variant of EEG was also described by other researchers [5,6]. Changes in the brain during magnetic resonance imaging in patients with pyridoxine-dependent epilepsy may vary from normal to diffuse atrophy of the gray and white matter of the hemispheres [2]; in our case no changes were detected.
According to Plecko B. Et al., with late diagnosis stable remission after the appointment of pyridoxal-5'-phosphate is observed only in a few patients [1]. Early treatment is critical to prevent irreversible damage to the central nervous system and shows positive results [1,5,6]. Patients with pyridoxine-dependent epilepsy require lifelong supplementation with pyridoxal-5'-phosphate. Therapeutic doses of the drug vary from 15 to 30 mg/kg/day [1]. The daily requirement for vitamin B6 in infancy is 0.1–0.3 mg. Pyridoxal-5'-phosphate doses up to 500 mg/day are considered safe in children with classical vitamin B6 deficiency, but higher doses may cause reversible sensory and rare motor neuropathy [1], so total daily doses of pyridoxal-5'-phosphate, should not exceed 200-300 mg. There are no data on the optimal dose of the vitamin for long-term treatment. In experimental animals, doses of pyridoxal 5'-phosphate >50mg/kg/d induce ataxia, peripheral neuropathy, and muscle weakness; histological examination demonstrates neuronal damage with loss of myelin and degeneration of sensory fibers in peripheral nerves, dorsal columns of the spinal cord, and descending tract of the trigeminal nerve. In most cases of peripheral neuropathy, the total dose of pyridoxal 5'-phosphate is >1000 mg/day. Some children who take high concentrations of pyridoxal-5'-phosphate develop a persistent increase in transaminases with progression to cirrhosis and hepatocellular carcinoma [3]. To avoid side effects, a fixed effective dose should be used. However, studies showed that daily doses up to 1100 mg/day and 50 mg/kg/day to achieve a state without epileptic seizures did not cause any side effects when they were divided into 4–5 doses per day [12]. In our case the doses of pyridoxal-5'-phosphate less than 600 mg/day induces epileptic seizures and cognitive disfunction. Some mutations in the genes encoding of pyridoxamine-5-phosphate oxidase may require the combined treatment with pyridoxal-5'-phosphate and pyridoxine [12,13]. It is possible that such treatment will have a positive response in our patient as well.
Another interesting feature of this clinical case is an intolerance of the patient to many products: remission occurs only on a low-protein, low-fat diet with the exclusion of dairy, meat products and eggs. Similar dietary restrictions are observed in ALDH7A1 deficiency (antiquitin deficiency), which often accompanies PNPO gene mutation. In our case ALDH7A1 deficiency was excluded by exome sequencing [13,14]. However, a lysine-restricted diet can also be effective for homozygous mutations in the PNPO gene in some patients [14]. As an example of a diet, the recommendations of Koelker and Ross on glutaric aciduria type I can be used [15].
The patient also has a high content of vitamin B6 in plasma (775.0 nmol/l), which is typical response to an intake of pyridoxal-5'-phosphate (described levels of vitamin B6 in plasma: 400 nmol/l, 1060 nmol /l and 624 nmol/l) [12,18]. It is not known why some patients continue to have seizures even when taking high doses of pyridoxal-5'-phosphate, while others grow almost normally [1,7,19]. The long-term prognosis for this patient remains unclear. For our patient a clarifying genetic study with modification of pharmacological treatment and diet is required, considering that the girl does not tolerate protein hydrolysates and an unstable clinical remission only on a low-protein low-fat diet with the exclusion of dairy, meat products and eggs.
CONCLUSIONS
DNA diagnostics using the method of sequencing of exome regions of the genome is a key method for early verification of the diagnosis of epilepsy in newborns and young children, which in combination with the therapy can improve the prognosis.
The presence of heterozygous mutations in this clinical case suggests other metabolic deficits, which complicates the selection of treatment and requires additional examination of the exome.
To ensure stable remission, nutritional correction is required to compensate for deficient conditions during severe elimination measures, as well as the selection of the minimum sufficient dosage of pyridoxal-5'-phosphate in combination with pyridoxine hydrochloride.
Topical neuropsychological diagnostics and psychological correction based on intact higher mental functions makes the recovery of the patient possible.
Conflict of Interest: The authors of this article have confirmed that there are no conflicts of interest or financial support to report.
For more information: https://jmedcasereportsimages.org/about-us/
For more submission : https://jmedcasereportsimages.org/
#focal epilepsy#Pyridoxal 5#phosphate#vitamin B6#PNPO#vitamin B6-dependent epilepsy#neuropsychological diagnostics#DNA#aheterogeneous#alpha-aminoadipine#semialdehyde#Plotnikova I.A#jcrmhs
2 notes
·
View notes
Note
I haven’t finished reading your meta on wizard genetics but sounds very interesting so far! You said typical xx/yy genetic model don’t work for passing magic in a way makes sense but what about a mutation gene? Like albinism etc??
Thanks for your great question! (And soo sorry for all the asks I haven't answered yet... I find it a lot harder to talk about my writing and headcanons than science in a public space, but I swear I'll get to them!)
So, traits like OC albinism are autosomal recessive, which means that a mutated gene is passed down through recessive alleles (I used m for the recessive allele in my meta example). In order for the mutation to be expressed, someone needs to inherit two recessive copies of the gene, one from each parent. While these conditions might seem like random mutations because they're so uncommon, the mutation actually already exists in the gene pool. It just crops up when two people who carry the allele both pass that mutation on to their child.
Basically, what that means is that everyone with OC albinism is most likely a super-distant relative of the first-ever person to express the mutation. The mutation can skip generations and still linger in the gene pool because it's recessive, so someone can carry just one copy without expressing it. This isn't the case for dominant mutations, since "carriers" of those genes express the physical trait. (There are exceptions to this rule, but they don't really apply here...)
The model doesn't work for magic because dominant traits don't skip generations, yet two Muggles can still have a magical child (if Magic is a dominant trait, Muggles have two recessive alleles each). This essentially suggests that the Magic mutation continues to crop up randomly, which is statistically really weird! We have a lot of DNA; even among 7+ billion people, it's super rare for the same exact mutation to randomly occur twice.
It's almost like... there's some kind of external power affecting those odds... something that can't be explained by science... 🤔 🪄💫
I hope that was helpful!
2 notes
·
View notes
Note
there’s a good wikipedia page on “genetic studies on jews” that would satisfy some of your curiosity. from its “comparison to non-jewish populations” section: “Many genetic studies have demonstrated that most of the various Jewish ethnic divisions and Druze, Palestinians,[4][68][5][44] Bedouin,[68][5] Lebanese people and other Levantines cluster near one another genetically.”
yes, thank you!
some highlights:
Citing autosomal DNA studies, Nicholas Wade estimates that "Ashkenazic and Sephardic Jews have roughly 30 percent European ancestry, with most of the rest from the Middle East." He further noticed that "The two communities seem very similar to each other genetically, which is unexpected because they have been separated for so long." Concerning this relationship he points to Atzmon's conclusions that "the shared genetic elements suggest that members of any Jewish community are related to one another as closely as are fourth or fifth cousins in a large population, which is about 10 times higher than the relationship between two people chosen at random off the streets of New York City".[12]
30 percent is shockingly low! low enough that im like, a little doubtful. only 30% over 2000 years? thats crazy
also i THINK this means that making jokes about queen elizabeth being cousins with her husband is antisemitic. they were third cousins!
anyway theres a TON of controversy in this section, unsurprisingly
A study led by Harry Ostrer published on 11 June 2010, found close links between Ashkenazi, Sephardi, and Mizrahi Jews, and found them to be genetically distinct from non-Jews. In the study, DNA from the blood of 237 Jews and about 2,800 non-Jews was analyzed, and it was determined how closely related they were through IBD. Individuals within the Ashkenazi, Sephardi, and Mizrahi groups shared high levels of IBD, roughly equivalent to that of fourth or fifth cousins. All three groups shared many genetic features, suggesting a common origin dating back more than 2,000 years. The study did find that all three Jewish groups did show various signs of admixture with non Jews, with the genetic profiles of Ashkenazi Jews indicating between 30% and 60% admixture with Europeans, although they clustered more closely with Sephardi and Mizrahi Jews.[98]
30-60% seems very plausible to me
however i think this is all about relatedness to other modern population groups, when im more curious about relatedness to the ancient people there. are there just not that many samples? they can get neanderthal DNA, feels like it shouldnt be hard to get ancient canaanite DNA
4 notes
·
View notes
Text
Arabian horse: Weight, Height, Life Expectancy, Health Special info
The Arabian Horse is the oldest recorded breed of horse, with reliable documentation and pictorial representations spanning at least 2,000 years that place the breed’s development in the Middle East region1,2,3.
Paleogenomic evidence supports the contribution of ancient Persian ancestry during the early formation of the modern European horse breed around 1100-1300 YA4. The desert horse expanded further with the rise of nomadic Bedouin tribes, who valued these horses as cultural symbols, sources of wealth, and military resources.
Today, despite being surpassed in absolute numbers by the American Quarter Horse, the Arabian breed is still the most widespread worldwide5, with pedigree registries in at least 82 countries.
The modern Arabian horse has a unique conformational phenotype that includes a dish-shaped facial profile, wide-set eyes, arched neck, and high tail 6.
However, Arabian horses in photographs taken in the late 1800s and early 1900s often show less pronounced facial dishing and lower tail carriage2, suggesting that these traits may be under strong selection by modern Arabian breeders, especially for lines of horses. Which is mainly used for non-riding. Show competitions.
The Arabian horse is also renowned for its heat tolerance and athletic endurance, making the Arabian a popular breed for long-distance races, where they carry the rider’s weight over distances of up to 160 km in a winning time of 8 h 7. Analysis of Arabian endurance horses has shown that the predisposition to this type of athletic competition is a multi-genic trait7.
Arabian horses have been exported from their ancestral homeland for many centuries. However, exported populations typically had fewer founder animals, and as a result, may now have limited genetic diversity8.
Taken together with the above selection for constitutive phenotype, the potential for deleterious breeding in Arabians should be high, and it is not surprising that several important autosomal recessive inherited diseases have been identified in Arabians 9,10.
In contrast, the few studies of the diversity of Arabian horses in the Middle East have shown higher levels of diversity in these horses compared to the progeny of Arabians exported to other parts of the world11,12,13.
Despite the evidence of the antiquity of the Arabian breed, there is relatively little solid documentation for the various breeds and maternal lineages of Arabian horses that are maintained by Arabian horse fanciers and breeders. In fact, several molecular studies using mitochondrial DNA have failed to confirm traditional Arabian horse maternal ancestry transmitted through oral history14,15,16,17.
The influence of Arabian horses in ‘improving’ other horse breeds has been generally acknowledged among equestrians for over 100 years18. The best-documented example of such influence is in the thoroughbred pedigree, which has been maintained as a stud book since 179119.
In a pedigree-based analysis of Thoroughbred founder lines, Cunningham and colleagues found that three stallions were imported. England has been a major contributor to the modern-day Thoroughbred gene pool since the Middle East in the late 18th century.
The Godolphin Arabian (sometimes referred to as Barb), is estimated by pedigree analysis to contribute 13.53% to the modern gene pool, as well as the Darley Arabian and the Byerley Turk20.
Recently, however, analysis of horse Y chromosome haplotypes has shown that the Y haplotype of the “Darley Arabian” actually originated from the Turkoman horse, an ancient breed from the Middle East and Central Asia that resembles the Arabian horse, also an “Oriental” type breed21. This calls into question the role of the Arabian as the founder of the Thoroughbred breed and, more generally, its influence on other horse breeds.
This study documents the population structure in a large global sample of the Arabian breed of horse. We used equine single nucleotide polymorphism (SNP) arrays and whole-genome re-sequencing in a comprehensive genome-wide analysis of the genetic diversity of Arabian horses that explored the population structure and origins of this breed.
In addition, we applied genome-wide analysis to find Arabian-specific genomic regions that show characteristic signals of selective action, and therefore variation that may be important for the Arabian horse’s unique physical traits.
Weight: 800 to 1,000 pounds
Height: 14 hands (56 inches) to 16 hands (64 inches)
Body Type: Lithe, compact body; small, wedge-shaped head; dished facial profile, long, arched neck
Best For Experienced owners and riders
Life Expectancy: 25 – 30 years
Origin: Arabian peninsula
Colors: Bay, chestnut, black, gray
Breed characteristics
Arabian horses have clean, wedge-shaped heads, broad foreheads, large eyes, large nostrils, and small muzzles. Most show a distinctive concave or “dished” profile.
Many Arabians also have a slight forehead bulge between the eyes, called a jibah by the Bedouin, which adds extra sinus capacity, believed to have helped the Arabian horse in its native arid desert environment. Another breed characteristic is the arched neck, with a large, well-set windpipe set on a pure, clean throat.
This structure of pole and throttle was called Mitba or Mitbeh by the Bedouins. In the ideal Arabian, it is long, allowing flexibility in the bridle and room for the windpipe.
Other distinguishing features are a relatively long, level crop or hindquarter top and a naturally high tail carriage. The USEF breed standard requires Arabians to have solid bones and a standard correct equine conformation. Well-bred Arabians have deep, well-angulated hips and well-set shoulders.
Within the genus, there are variations. Some individuals have wider, more powerfully muscled hindquarters suitable for sharp bursts in events such as reining, while others have longer, leaner muscles better suited for long stretches of flatwork such as endurance riding or horse riding.
Most have a compact body with a short back. Arabians generally have dense, strong bones and good hoof walls. They are particularly noted for their endurance, and the breed’s superiority in endurance riding shows that well-bred Arabians are robust, strong horses with superior endurance.

3 notes
·
View notes
Text
youtube
Discovering My Autosomal DNA Results
Thomas's roots trace back to Europe, showcasing a rich tapestry of diverse ancestry. Dive into the specifics as we explore his Scandinavian, Central European, and Irish heritage. But the journey doesn't stop there; we also uncover connections to Eastern Europe, Greece, the Balkans, and even the Middle East.
#AncestryJourney#AutosomalDNAResults#EuropeanHeritage#GeneticHistory#FamilyRoots#Genealogy#GeneticAncestry#GeneticHeritage#HeritageJourney#AutosomalDNA#MiddleEastHeritage#DNAResults#AshkenaziJew#SephardicJew#DruzeCommunity#Mesopotamia#GeneticGroups#DiverseRoots#DNAJourney#GenealogyDiscovery#FamilyHistory#Youtube
0 notes
Last Seen Blogs

nrblog
Untitled

madisonhentosh
Blood, Sweat, and Tears

sparklingcid3r
Those were the best years of my life

mirandaaaaxo-blog
miranda aaaxo For you

banga718-blog
BANGA SHINE TFMG 900BLOCKENT