#Cytochrome c
Explore tagged Tumblr posts
Visit Tumblr Blog
Explore Tumblr blogs with no restrictions, modern design and the best experience.
Last Seen Tumblr Blogs





Fun Fact
Tumblr has a low social media market share in South America.
Text
Mitochondrial Dysfunction in Cardiovascular Disease
Introduction
Mitochondria are essential organelles responsible for the production of cellular energy in the form of adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS). The heart, due to its continuous contractile activity, has a high energy demand and is critically dependent on mitochondrial function for normal physiological and pathological processes. Mitochondrial dysfunction has emerged as a central mechanism in the pathogenesis of cardiovascular diseases (CVDs), including ischemic heart disease, heart failure, hypertension, and arrhythmias. This technical overview discusses the molecular mechanisms of mitochondrial dysfunction in cardiovascular disease, its impact on cellular and organ function, and the potential therapeutic strategies to mitigate mitochondrial-related pathophysiology in CVDs.
Mitochondrial Function in Cardiovascular Cells
Mitochondria are highly dynamic organelles that perform several key functions crucial for the health of cardiovascular cells. They are involved in:
ATP Production via Oxidative Phosphorylation: In the mitochondria, energy production is driven by the electron transport chain (ETC), which is composed of complex I-IV embedded in the inner mitochondrial membrane. Electrons derived from NADH and FADH2 produced during the citric acid cycle are transferred through these complexes to ultimately reduce oxygen to water at complex IV. This electron transfer drives proton pumps that create an electrochemical gradient (proton motive force) across the inner mitochondrial membrane, which is utilized by ATP synthase (complex V) to produce ATP.
Calcium Homeostasis: Mitochondria play a crucial role in buffering intracellular calcium concentrations. They take up calcium from the cytoplasm in response to cellular signaling and help maintain cellular homeostasis by storing calcium in the matrix and releasing it when required for cellular signaling. Dysregulation of mitochondrial calcium handling can lead to pathophysiological conditions such as mitochondrial permeability transition (MPT) and cell death.
Reactive Oxygen Species (ROS) Production: Mitochondria are the primary source of ROS due to the incomplete reduction of oxygen molecules during electron transport in the ETC. Under normal conditions, low levels of ROS act as signaling molecules. However, excessive ROS generation due to mitochondrial dysfunction can cause oxidative stress, which damages cellular components such as lipids, proteins, and mitochondrial DNA (mtDNA), contributing to the pathogenesis of cardiovascular diseases.
Apoptosis and Cell Death: Mitochondria are central regulators of apoptosis. The release of pro-apoptotic factors such as cytochrome c from the mitochondrial intermembrane space into the cytoplasm triggers caspase activation, leading to programmed cell death. Mitochondrial dysfunction in cardiovascular tissues can lead to inappropriate cell death, contributing to the progression of CVDs.
Molecular Mechanisms of Mitochondrial Dysfunction in Cardiovascular Disease
Mitochondrial dysfunction in cardiovascular disease can result from several factors, including oxidative damage, altered mitochondrial dynamics, mutations in mitochondrial DNA, and defects in mitochondrial signaling. Below are the primary molecular mechanisms contributing to mitochondrial dysfunction in cardiovascular pathologies:
1. Oxidative Stress and ROS Accumulation
Excessive ROS generation is a hallmark of mitochondrial dysfunction and a major contributor to cardiovascular disease progression. Under normal conditions, the ETC produces ROS as a byproduct of electron transfer; however, under pathological conditions such as ischemia, hypoxia, or heart failure, there is an increase in mitochondrial ROS production. This increase is due to the altered electron flow through the ETC, particularly at complex I and III, which results in the incomplete reduction of oxygen.
The accumulation of ROS causes oxidative damage to mitochondrial lipids, proteins, and mtDNA. For instance, lipid peroxidation of mitochondrial membranes leads to membrane destabilization and disruption of mitochondrial function. ROS also modify proteins involved in mitochondrial dynamics and bioenergetics, impairing the capacity of mitochondria to generate ATP. Furthermore, oxidative damage to mtDNA leads to mutations that compromise the mitochondrial respiratory chain complexes, creating a vicious cycle of mitochondrial dysfunction.
2. Mitochondrial Permeability Transition (MPT) and Calcium Overload
Mitochondrial permeability transition is a critical event in mitochondrial dysfunction. The opening of the mitochondrial permeability transition pore (mPTP) occurs when the inner mitochondrial membrane becomes permeable to ions and small molecules, disrupting the electrochemical gradient required for ATP production. Under pathological conditions such as ischemia-reperfusion injury, excessive ROS and calcium overload activate the mPTP, leading to mitochondrial swelling, loss of membrane potential, and the release of pro-apoptotic factors (e.g., cytochrome c), triggering cell death.
Calcium overload plays a significant role in mitochondrial dysfunction. During stress conditions like ischemia, excessive intracellular calcium is taken up by mitochondria, causing mitochondrial matrix expansion and rupture of the mitochondrial membrane. This exacerbates cellular injury and promotes cell death pathways in the myocardium, contributing to myocardial infarction and heart failure.
3. Mitochondrial Dynamics Dysregulation
Mitochondrial dynamics refer to the continuous processes of mitochondrial fusion and fission that maintain mitochondrial quality and function. In response to cellular stress, mitochondria can undergo fission to segregate damaged components or fusion to promote functional compensation. Mitochondrial fission is regulated by dynamin-related protein 1 (DRP1), while fusion is mediated by mitofusins (MFN1 and MFN2) and optic atrophy 1 (OPA1). In cardiovascular diseases, this dynamic balance is often disrupted, leading to mitochondrial fragmentation, reduced mitochondrial function, and increased susceptibility to apoptosis.
In heart failure, for example, the upregulation of DRP1 and downregulation of fusion proteins contribute to mitochondrial fragmentation, reduced ATP production, and elevated ROS levels. This dysfunction is exacerbated by altered signaling pathways, including those associated with autophagy (mitophagy), which is responsible for removing damaged mitochondria. Dysfunctional mitophagy further impairs mitochondrial quality control, worsening cardiac injury.
4. Mitochondrial DNA Mutations
Mitochondrial DNA is more prone to mutations than nuclear DNA due to its proximity to the ETC and lack of protective histones. In cardiovascular diseases, mutations in mtDNA contribute to defective mitochondrial function. For example, mutations in genes encoding subunits of the OXPHOS complexes (such as ATP6, ND1, or CYTB) lead to impaired ATP synthesis and defective mitochondrial bioenergetics, contributing to myocardial ischemia and heart failure.
Mitochondrial mutations may also affect the regulation of ROS production and the activation of apoptotic pathways, accelerating tissue damage and organ dysfunction.
Therapeutic Approaches Targeting Mitochondrial Dysfunction in Cardiovascular Disease
Given the critical role of mitochondria in cardiovascular disease, several therapeutic strategies have been developed to target mitochondrial dysfunction and restore normal mitochondrial function. These include:
1. Mitochondrial Antioxidants
Mitochondrial-targeted antioxidants, such as MitoQ, MitoTEMPO, and SkQ1, have been developed to specifically target ROS within mitochondria. These compounds aim to reduce oxidative stress, limit mitochondrial damage, and improve mitochondrial function. Clinical studies are ongoing to assess the efficacy of these antioxidants in reducing myocardial injury and improving outcomes in heart failure and ischemic heart disease.
2. Mitochondrial Biogenesis Activation
Stimulating mitochondrial biogenesis to increase the number of functional mitochondria is another potential therapeutic strategy. Activators of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), a key regulator of mitochondrial biogenesis, are being investigated as potential treatments for heart failure. Exercise training is a natural way to activate PGC-1α and increase mitochondrial function, which has been shown to improve cardiac outcomes in patients with heart failure.
3. MPTP Inhibition
Inhibitors of the mPTP, such as cyclosporine A, have been studied for their potential to prevent ischemia-reperfusion injury by inhibiting pore opening. By preserving mitochondrial integrity, these inhibitors may help reduce myocardial damage and improve survival after myocardial infarction.
4. Gene Therapy and Mitochondrial Transplantation
Gene therapy approaches, including the use of CRISPR/Cas9 to correct mitochondrial DNA mutations, hold promise in treating mitochondrial diseases. Additionally, mitochondrial transplantation, where healthy mitochondria are delivered to damaged cardiac cells, is an emerging area of research, with the potential to restore mitochondrial function and improve heart function in patients with severe myocardial injury.
Conclusion
Mitochondrial dysfunction plays a central role in the pathogenesis of cardiovascular diseases, contributing to impaired ATP production, increased ROS production, and cell death. Understanding the molecular mechanisms underlying mitochondrial dysfunction provides critical insights into the development of novel therapeutic strategies. Approaches targeting mitochondrial biogenesis, oxidative stress, mitochondrial dynamics, and mPTP inhibition offer promising avenues for the treatment of cardiovascular diseases and could lead to more effective management of conditions such as heart failure, ischemic heart disease, and hypertension. However, further research and clinical trials are needed to fully elucidate the potential of these therapeutic strategies in improving cardiovascular health.
#Mitochondria#Cardiovascular Disease (CVD)#Mitochondrial Dysfunction#Oxidative Phosphorylation#ATP Production#Reactive Oxygen Species (ROS)#Mitochondrial DNA (mtDNA)#Mitochondrial Permeability Transition (MPT)#Calcium Homeostasis#Heart Failure#Ischemic Heart Disease#Hypertension#Mitochondrial Dynamics#Mitochondrial Fission and Fusion#Mitochondrial Biogenesis#Mitochondrial Antioxidants#Mitochondrial Targeted Antioxidants#MPTP Inhibitors#Gene Therapy#Mitochondrial Transplantation#Electrochemical Gradient#Mitochondrial Fragmentation#Cytochrome C#Cell Death (Apoptosis)#Peroxisome Proliferator-Activated#Receptor Gamma Coactivator 1-alpha (PGC-1α)
0 notes
Text
Testing my bestie’s patience by not being able to remember the names of any movies or actors correctly ever and pantomiming what I mean until she guesses it
#I’m just like my momma#she does screenwriting/directing so it’s extra trying for her#I’m sorry bestie unfortunately my brain is soup and I can only remember malate dehydrogenase cytochrome oxidase C ubiquinone nicotinamide#etc etc
0 notes
Text
Guide to Life-Sustaining Nutrients: Copper [PREVIEW]
The following is a preview of a Patreon-exclusive newsletter Click the icon below to support Become Something New for the cost of just one cup of coffee per month for access to this and upcoming Patreon-only content. ☕📖🧠💪 Patreon Starting at the most vital level, copper is a mitochondrial cofactor, essential for cytochrome C oxidase (complex IV), which completes oxidative phosphorylation to…
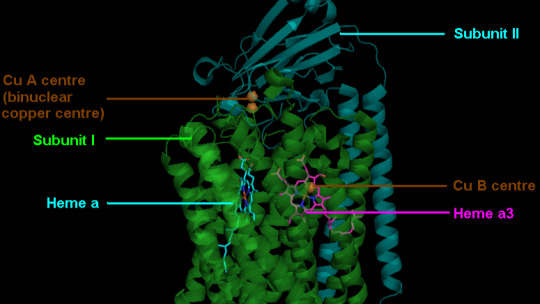
View On WordPress
#adrenaline#aging#allergies#alzheimer&039;s#Amyotrophic Lateral Sclerosis#apoptosis#arterial plaques#arthritis#aspartame#avocado#beauty#biohacking#blood pressure#bone health#cancer#cartilage#chris masterjohn#collagen#cytochrome c oxidase#dark chocolate#dementia#dhea#diabetes#diamine oxidase#disulfiram#dopamine#elastin#epinephrine#ferrous iron#gray hair
0 notes
Text

Convergent evolution of Amphidromus-like colourful arboreal snails and phylogenetic relationship of East Asian camaenids, with description of a new Aegistohadra species
Parin Jirapatrasilp, Chih-Wei Huang, Chung-Chi Hwang, Chirasak Sutcharit and Chi-Tse Lee
ABSTRACT
East Asian terrestrial snails of the family Camaenidae are diverse in terms of genus and species numbers, shell morphology and mode of living. This family also includes colourful conical arboreal snails that traditionally have been assigned to the genus Amphidromus. Yet, the present study shows that, despite their deceiving conchological similarity, some of these East Asian arboreal snails do not belong to the genus Amphidromus or the subfamily Camaeninae. The presence of a dart complex comprising a mucous gland, a dart sac, an accessory sac and a proximal accessory sac, along with a pronounced penial caecum and molecular phylogenetic analyses revealed that former ‘Amphidromus’ dautzenbergi, ‘A.’ roemeri and ‘Camaena’ mirifica, and one additional new species belong to Aegistohadra (subfamily Bradybaeninae). Aegistohadra dautzenbergi, comb. nov. and Aegistohadra roemeri, comb. nov. are conical with colourful spiral bands, whereas Aegistohadra mirifica, comb. nov. and Aegistohadra zhangdanae, sp. nov. are heliciform to conical with colourful, variegated spiral and transverse banding patterns. DNA sequence analyses also revealed that each variety of Aegistohadra dautzenbergi could not be differentiated by mitochondrial (cytochrome c oxidase subunit I and 16S rRNA) gene fragments. The phylogenetic position of Aegistohadra within the East Asian camaenids revealed that the similar appearance in shell morphology, microhabitat use and diet to arboreal snails in the genus Amphidromus is homoplastic. Moreover, the presence or absence of a dart complex is also homoplastic and is unsuitable for suprageneric classification. By contrast, the presence of a flagellum and a penial caecum is useful for the suprageneric classification.
Read the paper here:
CSIRO PUBLISHING | Invertebrate Systematics
#tree snail#land snail#snail#gastropod#mollusk#amphidromus#camaenidae#animals#nature#asia#malacology
111 notes
·
View notes
Text
Call me a DNA barcode the way you compare me to a reference sample of a 600bp region of the mitochondrial cytochrome oxidase c I gene
39 notes
·
View notes
Text

Yesssss new tiny knifefish species described from Brazil, everyone say hello Microsternarchus javieri
Shamelessly copying and pasting from the Fish In The News facebook page:
𝑀𝑖𝑐𝑟𝑜𝑠𝑡𝑒𝑟𝑛𝑎𝑟𝑐ℎ𝑢𝑠 𝑗𝑎𝑣𝑖𝑒𝑟𝑖, a new species of weakly electric knifefish is described from the flooded savanna streams of the Branco River and in terra-firme streams in the mid-and lower portions of the Negro River basin, Amazonas, Brazil.
Open-access - https://www.scielo.br/j/aa/a/4hGwQYNxjyGgpQP5XJF73Ft/
𝗥𝗲𝘀𝗲𝗮𝗿𝗰𝗵 𝗧𝗶𝘁𝗹𝗲
𝑀𝑖𝑐𝑟𝑜𝑠𝑡𝑒𝑟𝑛𝑎𝑟𝑐ℎ𝑢𝑠 𝑗𝑎𝑣𝑖𝑒𝑟𝑖, a new species of weakly electric fish (Gymnotiformes: Hypopomidae, Microsternarchini) from the Negro River basin, Amazonas, Brazil
𝗖𝗶𝘁𝗮𝘁𝗶𝗼𝗻
ESCAMILLA PINILLA, C., COX FERNANDES, C., & ALVES-GOMES, J. A.. (2025). 𝑀𝑖𝑐𝑟𝑜𝑠𝑡𝑒𝑟𝑛𝑎𝑟𝑐ℎ𝑢𝑠 𝑗𝑎𝑣𝑖𝑒𝑟𝑖, a new species of weakly electric fish (Gymnotiformes: Hypopomidae, Microsternarchini) from the Negro River basin, Amazonas, Brazil. Acta Amazonica, 55, e55bc24175. https://doi.org/10.1590/1809-4392202401751
𝗔𝗯𝘀𝘁𝗿𝗮𝗰𝘁
Here we describe a new hypopomid species, 𝑀𝑖𝑐𝑟𝑜𝑠𝑡𝑒𝑟𝑛𝑎𝑟𝑐ℎ𝑢𝑠 𝑗𝑎𝑣𝑖𝑒𝑟𝑖i n. sp., encountered in flooded savanna streams of the Branco River and in terra-firme streams in the mid-and lower portions of the Negro River basin. We compared this new species with 𝑀. 𝑏𝑖𝑙𝑖𝑛𝑒𝑎𝑡𝑢𝑠 from the San Bartolo River, Venezuela, and M. brevis from the upper portion of the Negro River.
We also compared this new species with two recently described species in the genus 𝑀𝑖𝑐𝑟𝑜𝑠𝑡𝑒𝑟𝑛𝑎𝑟𝑐ℎ𝑢𝑠, 𝑀. 𝑙𝑜𝑛𝑔𝑖𝑐𝑎𝑢𝑑𝑎𝑡𝑢𝑠 and 𝑀. 𝑠𝑐ℎ𝑜𝑛𝑚𝑎𝑛𝑛𝑖. We examined morphometrics, anatomical characters, DNA barcode distances for the COI (cytochrome C oxidase subunit I) gene, and electric organ discharge (EOD) parameters. We diagnosed 𝑀. 𝑗𝑎𝑣𝑖𝑒𝑟𝑖 n.sp. based on variation in maximum body depth, eye diameter, caudal vertebral counts, number of anal fin rays, and the shape of the maxillae. The average intra-specific genetic distance (K2P) in 𝑀. 𝑗𝑎𝑣𝑖𝑒𝑟𝑖 n.sp. was 0.83%, whereas the average inter-specific genetic distance to 𝑀. 𝑏𝑟𝑒𝑣𝑖𝑠 was 12.45%, and to other hypopomids ranged from 17.21 to 21.54%. When comparing EOD waveforms of the new species with 𝑀. 𝑏𝑟𝑒𝑣𝑖𝑠, we found differences in repetition rate, the ratio between the first and second phase areas, and the polarity balance.
The description of 𝑀. 𝑗𝑎𝑣𝑖𝑒𝑟𝑖 n. sp. increases to five the number of species in the genus.
𝗘𝘁𝘆𝗺𝗼𝗹𝗼𝗴𝘆
The specific epithet, 𝑗𝑎𝑣𝑖𝑒𝑟𝑖, is in honor of the late Javier Maldonado Ocampo, whose research on gymnotiforms, systematics, and conservation greatly contributed to our understanding of the Neotropical ichthyofauna. A masculine noun in apposition.
𝗣𝗵𝗼𝘁𝗼 𝗖𝗿𝗲𝗱𝗶𝘁
Lateral view of 𝑀𝑖𝑐𝑟𝑜𝑠𝑡𝑒𝑟𝑛𝑎𝑟𝑐ℎ𝑢𝑠 𝑗𝑎𝑣𝑖𝑒𝑟𝑖 n. sp. from the Negro River basin. A - Holotype (INPA-ICT 060886), 98.6 mm TL, 67.6 mm LEA; B - Paratype (ANSP 212283), 101.2 mm TL, 62.0 mm LEA; C -Non-type (INPA-ICT 28591), 86.9 mm TL, 60.4 mm LEA live specimen.
Copyright © 2025 the Author(s). Published in Acta Amazonica journal. This paper is released under a Creative Commons Attribution 4.0 International (CC BY 4.0) licence. https://creativecommons.org/licenses/by/4.0/
#NewSpeciesAlert #NewSpecies #Ichthyology #Ictiology #Taxonomy #Biodiversity #Aquarium #AquariumHobby #Fishkeeping #Fishkeeper #Aquarist #Knifefish #ElectricFishes #ElectricFish #WeaklyElectricFish #Microsternarchus #Gymnotiformes #Hypopomidae #Microsternarchini #Amazonas #Neotropical #NeotropicalFishes #NegroRiver #RioNegro #Gymnotiforms #Systematics
36 notes
·
View notes
Text
Sup hi here's Red Bull tierlist


Left one is Dewdrop's and right is mine. We've only included flavors we've actively tried (besides coconut since we have it in the fridge and will reblog this post with a ranking for it when we try it)
[ Tagging: @cytochrome-sea since you expressed interest in seeing this ^^ ]
Text version of tier lists under cut
Tierlist for Dewdrop
S tier: Amber
A tier: blue and red
B tier: sugar free and original
C tier is empty
D tier: Yellow
F tier: Pink
My tierlist
S tier: amber and blue
A tier: red
B and C tier are empty
D tier: Yellow, sugar free and original
F tier: pink
12 notes
·
View notes
Text
Cyanide Poison

Let's start by understanding exactly how cyanide kills you. In simple terms, cyanide prevents cells from using oxygen to make energy molecules.
The cyanide ion, CN-, binds to the iron atom in cytochrome C oxidase in the mitochondria of cells. It acts as an irreversible enzyme inhibitor, preventing cytochrome C oxidase from doing its job, which is to transport electrons to oxygen in the electron transport chain of aerobic cellular respiration. Now unable to use oxygen, the mitochondria can't produce the energy carrier adenosine triphosphate (ATP). Tissues that require this form of energy, such as heart, muscle cells, and nerve cells, quickly expend all their energy and start to die. When a large enough number of critical cells die, you expire as well. Death usually results from respiratory or heart failure.
Immediate aymptoms include headaches, nausea and vomiting, dizziness, lack of coordination, and rapid heart rate. Long exposure symptoms include unconsciousness, convulsions, respiratory failure, coma and death.
A person exposed to cyanide may have cherry-red skin from high oxygen levels, or dark blue coloring, from Prussian blue (iron-binding to the cyanide ion). In addition to this, skin and body fluids may give off an almond odor.
The antidotes for cyanide include sodium nitrite, hydroxocobalamin, and sodium thiosulfate.
A high dose of inhaled cyanide is lethal too quickly for any treatment to take effect, but ingested cyanide or lower doses of inhaled cyanide may be countered by administering antidotes that detoxify cyanide or bind to it. For example, hydroxocobalamin, natural vitamin B12, reacts with cyanide to form cyanocobalamin, which leaves the body in urine.
These antidotes are administrated via injection, or IV infusion.
Cyanide is actually a lot more common than you'd think. It's in pesticides, fumigants, plastics, and electroplating, among other things. However, not all cyanide are so poisonous. Sodium cyanide (NaCN), potassium cyanide (KCN), hydrogen cyanide (HCN), and cyanogen chloride (CNCl) are lethal, but thousands of compounds called nitriles contain the cyanide group, yet aren't as toxic. They still aren't terribly good for you, so I wouldn't go around ingesting other cyanide compounds, but they're not quite as dangerous as the lethal kind.
Thank you for reading, have a lovely day :)
#cyanide#poison#cyanide poison#tw poison#poisons#chemistry#?#if it counts lmao#crime#criminal#investigation#forensics#scienceblr#science#sherlock#sherlock holmes
49 notes
·
View notes
Text
Meanwhile, at the Annual Mollusk Taxonomy Convention
Taxonomist 1: I think this population of blorb snails count as their own species under the phylogenetic and biological species concepts. Their last common ancestor was between 5 and 100 million years ago, idk, my 35 year old copy of Clustal finally exploded so I just eyeballed it, but they totally got wack cytochrome C oxidase genes, even though for the most part they're genetically pretty similar and they still look and act exactly the same in every way and also both live in the same place as each other. The only reason why they don’t interbreed is they have like a single incompatible protein thingo, which is also more or less the only meaningful phenotypic difference between them.
(this is an actual thing that can happen. They’re called cryptic species and species complexes and they hurt my soul)
Taxonomist 2: No. It’s more pragmatic and useful to just use the morphological and ecological species concepts here; and they say fuck you and your dumb snails. I wanna lump all existing species into half as many species, there’s too many fucking species.
Taxonomist 1: you wanna fucking say that to my face you little shit?
Taxonomist 3: Hey, real quick, what do you guys think about the possibility of reclassifying the Blorb genus under Littorinidae instead of Muricidae? Because I already wrote a paper on it, so that's the case now. Cry about it.
Taxonomist 2: I think today is the day bitches die.
*Mexican standoff using conch shells as blunt weapons ensued, there were no survivors.*
*This is unfortunately the leading cause of death among all taxonomists*
#science#biology#taxonomy#evolution#marine life#scientists sitcom#roughly based on a totally true story from my professor. He is a veteran of the mollusk taxonomy wars#No you cannot actually just 'eyeball' it but you would not believe some of the ancient software we be usin' in taxonomy.#sea snails#mollusk#blorbsday
82 notes
·
View notes
Text
The Connection Between Damaged Mitochondria and Arthritis

Mitochondria are integral organelles responsible for various critical cellular functions, primarily energy production through oxidative phosphorylation. They are involved in maintaining cellular homeostasis, regulating metabolism, modulating calcium levels, and controlling apoptosis. Emerging evidence has highlighted mitochondrial dysfunction as a key contributor to a variety of diseases, including arthritis. This formal overview aims to explore the complex relationship between damaged mitochondria and arthritis, focusing on the molecular mechanisms that link mitochondrial dysfunction to the pathogenesis of inflammatory joint diseases, particularly rheumatoid arthritis (RA) and osteoarthritis (OA).
Mitochondrial Structure and Function
Mitochondria are double-membraned organelles found in eukaryotic cells, and they are crucial for cellular energy metabolism. Their primary role is the production of adenosine triphosphate (ATP) via oxidative phosphorylation, a process that takes place in the inner mitochondrial membrane. During this process, the electron transport chain (ETC) generates a proton gradient across the inner membrane, which drives ATP synthesis through ATP synthase. However, this process also generates reactive oxygen species (ROS) as byproducts, primarily from complexes I and III of the ETC. Under normal physiological conditions, ROS are neutralized by antioxidants, including superoxide dismutase (SOD), catalase, and glutathione. However, under pathological conditions, excessive ROS production can lead to oxidative stress, contributing to cellular damage and dysfunction.
In addition to ATP production, mitochondria have essential roles in calcium buffering, apoptosis regulation, and the maintenance of cellular integrity. Damage to these organelles disrupts these functions, contributing to various diseases, including arthritis.
Mitochondrial Dysfunction in Arthritis
Arthritis is a group of diseases characterized by inflammation and degeneration of the joints. It includes conditions like rheumatoid arthritis (RA), an autoimmune disease, and osteoarthritis (OA), a degenerative disease. In both types of arthritis, mitochondrial dysfunction has been identified as a critical factor that exacerbates disease progression through several mechanisms, including increased oxidative stress, immune activation, and tissue damage.
1. Oxidative Stress and Mitochondrial Damage
Oxidative stress is a hallmark of both RA and OA, and mitochondria are central to its production. In these conditions, mitochondrial dysfunction results in an increase in ROS production, overwhelming the cell’s antioxidant defenses. This oxidative stress leads to the modification of cellular structures, including proteins, lipids, and DNA, causing further mitochondrial damage. In RA, pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1 (IL-1), and interleukin-6 (IL-6) stimulate immune cells like macrophages and neutrophils to release large amounts of ROS. These ROS contribute to the local inflammatory environment and accelerate joint destruction by damaging mitochondria and amplifying oxidative stress.
Mitochondrial damage results in a feedback loop where impaired mitochondrial function generates more ROS, further promoting inflammation. For instance, in RA, markers of oxidative damage such as 8-hydroxy-2'-deoxyguanosine (8-OHdG) and malondialdehyde (MDA) have been found to correlate with disease activity, suggesting a direct relationship between mitochondrial dysfunction and disease severity.
2. Mitochondrial DNA Damage and Inflammatory Signaling
Mitochondrial DNA (mtDNA) is particularly vulnerable to oxidative damage due to its proximity to the ETC, where ROS are produced during ATP synthesis. Unlike nuclear DNA, mtDNA is not protected by histones and has limited repair mechanisms, making it prone to mutations. Damage to mtDNA impairs mitochondrial function and can lead to the release of mtDNA fragments into the cytoplasm or extracellular space.
In the context of arthritis, mtDNA damage has been implicated in immune activation. When damaged mtDNA is released into the cytoplasm, it is recognized by pattern recognition receptors (PRRs), such as toll-like receptors (TLRs), on immune cells. TLRs, particularly TLR9, activate downstream inflammatory signaling pathways that lead to the production of pro-inflammatory cytokines such as TNF-α and IL-6. This further exacerbates the inflammatory response in joints and contributes to the progression of arthritis. Studies have shown that the presence of mtDNA fragments in the serum of RA patients correlates with disease activity, indicating the role of mtDNA in driving inflammation.
3. Mitochondrial Dynamics and Arthritis Pathogenesis
Mitochondrial dynamics refer to the continuous processes of mitochondrial fission (division) and fusion (joining), which maintain mitochondrial function and integrity. Fission allows for the removal of damaged mitochondria, while fusion helps to integrate mitochondrial contents and maintain a healthy mitochondrial pool. Imbalance between fission and fusion is associated with several diseases, including arthritis.
In the case of RA, excessive mitochondrial fission and reduced fusion have been observed. This imbalance results in mitochondrial fragmentation, which impairs mitochondrial function, increases ROS production, and contributes to cellular stress. Fission is regulated by proteins such as dynamin-related protein 1 (Drp1) and fission 1 protein (Fis1), while fusion is controlled by mitofusins (Mfn1 and Mfn2) and optic atrophy 1 (OPA1). Dysregulation of these proteins in RA leads to a fragmented mitochondrial network, which exacerbates oxidative stress and inflammation in synovial tissues.
4. Mitochondrial-Dependent Cell Death
Mitochondria are also central regulators of programmed cell death, particularly apoptosis. In the pathogenesis of arthritis, excessive or dysregulated apoptosis contributes to joint destruction. Mitochondrial dysfunction plays a critical role in the intrinsic apoptotic pathway by releasing pro-apoptotic factors such as cytochrome c and apoptosis-inducing factor (AIF). These factors activate caspase-dependent and caspase-independent pathways, leading to the death of synovial cells and cartilage cells, which contributes to the progressive tissue damage observed in both RA and OA.
Furthermore, mitochondrial permeability transition pore (mPTP) opening, which is induced by oxidative stress, can lead to necrosis, a form of uncontrolled cell death. Necrotic cell death in the joints increases inflammation and tissue degradation, particularly in OA, where cartilage breakdown is a hallmark feature.
Therapeutic Approaches Targeting Mitochondrial Dysfunction in Arthritis
Given the significant role of mitochondrial dysfunction in the pathogenesis of arthritis, various therapeutic strategies aimed at improving mitochondrial function are under investigation.
1. Mitochondrial Antioxidants
Mitochondrial-targeted antioxidants, such as MitoQ and MitoTEMPO, have been developed to selectively accumulate in mitochondria, where they can neutralize ROS and reduce oxidative stress. These compounds have shown promise in preclinical models of arthritis, where they help to reduce inflammation, protect mitochondrial function, and limit joint damage. The use of mitochondrial antioxidants could be an effective strategy to mitigate oxidative stress in arthritic conditions.
2. Mitochondrial Biogenesis Enhancement
Another potential therapeutic approach is the activation of mitochondrial biogenesis, the process by which new mitochondria are formed to compensate for damaged mitochondria. Agents that activate peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), a key regulator of mitochondrial biogenesis, could help restore mitochondrial function in arthritic tissues. Compounds such as resveratrol and NAD+ precursors are under investigation for their ability to promote mitochondrial biogenesis and improve cellular metabolism in arthritis.
3. Mitochondrial Dynamics Modulation
Restoring the balance between mitochondrial fission and fusion is another therapeutic strategy. Inhibiting excessive mitochondrial fission or promoting mitochondrial fusion may help maintain mitochondrial integrity and reduce inflammation in arthritis. Drugs targeting Drp1 or enhancing Mfn1/Mfn2 activity are potential candidates for modulating mitochondrial dynamics in arthritic diseases.
4. Mitophagy Enhancement
Mitophagy, the selective autophagic degradation of damaged mitochondria, is essential for maintaining mitochondrial quality. Enhancing mitophagy through the use of compounds like spermidine or activators of the PINK1/PARK2 pathway could help eliminate dysfunctional mitochondria and reduce inflammation, making it a promising therapeutic approach in arthritis.
Conclusion
Mitochondrial dysfunction plays a critical role in the pathogenesis of arthritis, contributing to oxidative stress, inflammation, and joint damage. The intricate relationship between damaged mitochondria and immune activation highlights the importance of targeting mitochondrial health in the treatment of arthritis. Emerging therapeutic strategies aimed at restoring mitochondrial function, reducing oxidative stress, and modulating mitochondrial dynamics hold promise for improving the management of arthritis and preventing joint destruction. Further research into mitochondrial biology and its role in arthritis is essential for the development of more effective, targeted therapies for these debilitating conditions.
#Mitochondrial dysfunction#Autoimmune disorders#Oxidative stress#Reactive oxygen species (ROS)#Mitochondrial dynamics#Mitochondrial fission#Mitochondrial fusion#Mitophagy#Apoptosis#Mitochondrial DNA (mtDNA)#Damage-associated molecular patterns (DAMPs)#Immune cell activation#Systemic lupus erythematosus (SLE)#Rheumatoid arthritis (RA)#Multiple sclerosis (MS)#Pattern recognition receptors (PRRs)#Toll-like receptors (TLRs)#Pro-inflammatory cytokines#Cytochrome c#NF-κB signaling#MitoQ#MitoTEMPO#Spermidine#PINK1/PARK2 pathway#Mitochondrial-targeted antioxidants#Immune dysregulation#Chronic inflammation#Mitochondrial fragmentation#Mitochondrial permeability transition pore (mPTP)#Autoantibodies
0 notes
Text
Story at-a-glance
Suppression of mitochondrial ATP production prevents apoptosis and activates the NLRP3 inflammasome, a key player in inflammation and disease
Inhibitors of oxidative phosphorylation (OXPHOS) lead to changes in mitochondrial cristae structure and retention of cytochrome c, which is necessary for NLRP3 activation but not sufficient on its own
Activation of the NLRP3 inflammasome requires two signals, one of which is mitochondrial, highlighting the complexity of its regulation
Diverse NLRP3 activators share the ability to suppress apoptosis, allowing damaged cells to survive and contributing to chronic inflammation and cancer
Mitochondrial dysfunction is closely linked to inflammation and various diseases, emphasizing the importance of understanding these mechanisms for optimal health
7 notes
·
View notes
Text
Unraveling the Tapestry of Cellular Energy: A Comprehensive Voyage through the Electron Transport Chain 🧬⚙️
Prepare for a deep dive into the labyrinthine pathways of the Electron Transport Chain (ETC), where molecular machinations weave the intricate tapestry of cellular respiration. In this odyssey, we'll navigate the complexities with surgical precision, leaving no nuance unexplored.
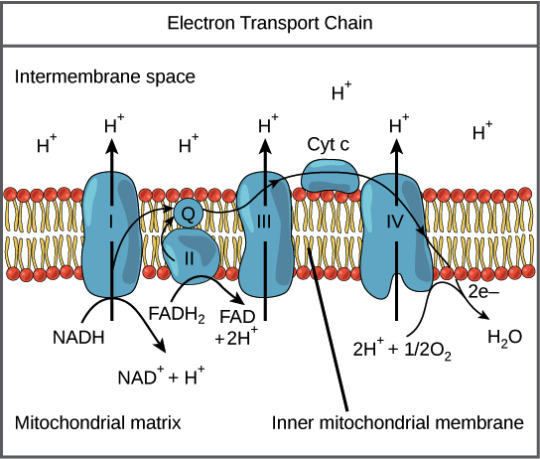
1. Prelude at Complex I (NADH Dehydrogenase):
The ETC's overture commences at Complex I, where NADH, a product of glycolysis and the Krebs cycle, surrenders its high-energy electrons. Traverse the serpentine route of flavin mononucleotide (FMN) and a succession of iron-sulfur clusters, witnessing the orchestrated dance that propels electrons toward the enigmatic ubiquinone (Q).
2. Interlude with Succinate (Complex II - Succinate Dehydrogenase):
As the symphony progresses, Complex II takes the stage with succinate as its protagonist. Succinate dehydrogenase, fueled by succinate from the Krebs cycle, orchestrates a parallel electron flow. Behold the ballet of electrons navigating iron-sulfur clusters and flavin adenine dinucleotide (FAD), converging upon ubiquinone (Q) in a seamless choreography.
3. Cytochrome Waltz (Complex III - Cytochrome bc1 Complex):
The narrative crescendos at Complex III, the cytochrome bc1 complex, where Q takes center stage. Through a series of mesmerizing redox reactions, Q gracefully shuttles electrons to cytochrome c. This transient dancer becomes the ethereal messenger, ferrying electrons with finesse towards the climactic rendezvous at Complex IV.
4. Grand Finale with Complex IV (Cytochrome c Oxidase):
In the climactic finale, Complex IV, personified by cytochrome c oxidase, awaits the electron ensemble. Watch as electrons, guided by a cascade of copper and iron centers, engage in a captivating pas de deux with molecular oxygen. Witness the alchemical metamorphosis as oxygen is humbly transmuted into water, marking the zenith of our electron saga.
5. Proton Symphony and ATP Synthesis:
Simultaneously, the proton symphony unfolds as protons, displaced during electron transit, accumulate in the intermembrane space. This sets the stage for a grand energy transfer. The finale crescendos with protons flowing back through ATP synthase, a molecular turbine, culminating in the synthesis of ATP—the lifeblood of cellular energy currency.
References:
1. Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K., & Walter, P. (2014). Molecular Biology of the Cell (6th ed.). Garland Science.
2. Nelson, D. L., Cox, M. M. (2017). Lehninger Principles of Biochemistry (7th ed.). W.H. Freeman and Company.
3. Berg, J. M., Tymoczko, J. L., Gatto, G. J. S., & Stryer, L. (2019). Biochemistry (8th ed.). W.H. Freeman and Company.
#science#biology#college#education#school#student#medicine#doctors#health#healthcare#molecules#chemistry#molecular biology
55 notes
·
View notes
Text
Oh, okay. The thing I need to do to induce P-cytochrome enzymatic action is: start smoking. No! Fuck off!
#is this why dad and i (only nonsmokers in the family) constantly OVERREACT to medications#while mom and my siblings UNDERREACT#medical cw#smoking cw#tobacco cw#nicotine cw#be careful 3liza#p cytochrome
6 notes
·
View notes
Text
POR Protein-GST Fusion
POR Protein-GST Fusion Catalog number: B2024072 Lot number: Batch Dependent Expiration Date: Batch dependent Amount: 20 ug Molecular Weight or Concentration: N/A Supplied as: Solution Applications: a molecular tool for various biochemical applications Storage: –70°C Keywords: CYPOR, NADPH–cytochrome P450 reductase, P450R Grade: Biotechnology grade. All products are highly pure. All solutions are…
0 notes
Text
0 notes
Text
Photobiomodulation: A Non-Invasive Solution for Low Back Pain
Low back pain (LBP) is more than just a discomfort—it's a global health concern that affects millions of people and often leads to long-term disability, limited mobility, and a lower quality of life. From office workers with sedentary routines to athletes dealing with injuries, low back pain doesn’t discriminate. Traditional treatments often involve medications, physical therapy, or in severe cases, surgical interventions. However, a promising, non-invasive therapy is gaining ground: Photobiomodulation (PBM).

A New Era in Pain Relief
In 2015, I had the privilege of speaking with Harvard researcher Dr. Michael Hamblin, one of the world’s most cited experts in Photobiomodulation. Dr. Hamblin has spent over three decades exploring how light therapy affects cellular health and how it could be harnessed to support recovery in conditions ranging from chronic pain to brain injuries.
During our conversation, Dr. Hamblin emphasized a particularly exciting finding: PBM’s anti-inflammatory effects, which are now being widely recognized as a breakthrough in the field of chronic pain management. He generously shared one of his most cited studies with me, which clearly outlines how PBM works at a cellular level to reduce inflammation and support healing—especially relevant for low back pain sufferers.
What is Photobiomodulation?
Photobiomodulation, also known as low-level laser therapy (LLLT) or light therapy, involves the application of red and near-infrared light to specific areas of the body. The goal is to stimulate biological processes at a cellular level—primarily through the mitochondria, the powerhouse of the cell.
PBM enhances mitochondrial function by activating cytochrome c oxidase, which leads to increased ATP (cellular energy) production, modulation of reactive oxygen species, and improved nitric oxide release. This chain of effects contributes to:
Accelerated tissue repair
Reduced inflammation
Improved circulation
Decreased pain sensitivity
Unlike heat-based or ablative lasers, PBM uses light wavelengths that do not damage tissue but instead support the body’s natural healing mechanisms.
How PBM Helps in Low Back Pain
Chronic low back pain often stems from inflammation, muscular strain, or disc issues. PBM addresses these challenges in a holistic, targeted way—without relying on medications or surgical procedures.
✳ Key Benefits of PBM in LBP Treatment:
Anti-Inflammatory Action: PBM modulates cytokines like TNF-α and IL-6, key players in the body’s inflammatory response. By reducing these pro-inflammatory signals, PBM can ease stiffness, swelling, and chronic pain in the lower back.
Clinical Results: A growing body of clinical evidence supports PBM’s effectiveness. Randomized controlled trials have shown that PBM significantly reduces pain and disability in people with chronic non-specific low back pain when compared to placebo treatments.
Minimal Side Effects: Unlike long-term medication use, PBM doesn’t come with side effects like dependency, gastrointestinal discomfort, or systemic risks. It’s safe, non-invasive, and can be applied repeatedly over time.
A Timely Alternative to Opioids and Surgery
The global opioid crisis has highlighted the need for safer alternatives in chronic pain management. Many conventional treatments only mask symptoms temporarily or bring a host of side effects. In contrast, PBM works by addressing the root causes of pain—tissue inflammation, cell damage, and poor circulation.
Integrating Photobiomodulation into a broader pain management plan may reduce the reliance on pharmaceutical interventions and improve long-term outcomes. Patients dealing with postural strain, injury recovery, degenerative disc disease, or other spine-related issues can find relief and restoration without resorting to addictive painkillers or high-risk surgeries.
Personalizing Pain Relief: PBM in Clinical Practice
One of the strengths of PBM is its adaptability. It can be administered in a variety of healthcare settings—from chiropractic and physiotherapy clinics to specialized pain centers. Practitioners often tailor the light wavelength, intensity, and duration based on a patient’s specific condition and severity of pain.
This precision allows for personalized care, with the ability to treat the affected areas directly without affecting other parts of the body. Sessions are typically quick and painless, often lasting just 10–20 minutes, with no downtime required afterward.
Looking Ahead: The Future of PBM in Pain Medicine
As more research supports the efficacy of PBM in musculoskeletal and neurological disorders, it’s expected to become a mainstay in chronic pain treatment. New clinical protocols and improved device accessibility mean patients may soon see this therapy integrated more widely into mainstream healthcare.
In addition to back pain, PBM is also being explored for conditions such as:
Neuropathic pain
Arthritis and joint pain
Sports injuries
Post-surgical recovery
Concussions and brain trauma
The possibilities are expanding—and the potential is profound.
Final Thoughts
Photobiomodulation represents a safe, science-backed, and natural way to manage low back pain. For those struggling with chronic discomfort and looking for options beyond pills and surgery, PBM provides real hope for healing. It targets the biological roots of pain—not just the symptoms—and empowers your body’s ability to recover.
Whether you’re an athlete, an office worker, or simply someone dealing with the wear and tear of daily life, Photobiomodulation may be the non-invasive answer you’ve been searching.
#advanced laser therapy#southwest pain relief#pain relief clinic#laser therapy#chatham pain clinic#treatment#quick start pain relief
0 notes